The selenocysteine insertion sequence in E. coli
© Anders Skovly 2025
As described in the article on translation in bacteria, the amino acid selenocysteine is incorporated into proteins through a unique mechanism that is not shared by any of the other amino acids. This article presents select methods and results from seven research texts focused on this mechanism. It is very much a niche topic, and probably of interest mainly for those who perform genetic engineering with genes containing selenocysteine codons. However, the article could still be of interest for others, as it describes a number of molecular techniques.
Zinoni, Heider, Bock (1990): Features of the formate dehydrogenase mRNA necessary for decoding of the UGA codon as selenocysteine
Most UGA-codons function as stop codons, but a few UGA-codons instead function as selenocysteine-codons. These few UGA don't bind to RF2 (Release factor 2), but to tRNASecACU (the anticodon is written from the 3'-end towards the 5'-end). One example is E. coli's fdhF gene, where codon number 140 is a UGA which is translated to selenocysteine.
The binding of tRNASecACU to UGA is dependent on a protein called SelB. This in contrast to every other elongator-tRNA, which depends on the protein EF-Tu (Elongation factor Tu) to bind their codons. SelB and EF-Tu have a large similarity in their amino acid sequences, but SelB contains an "extra part" (extra amino acid sequence) at one end of the chain which is not present in EF-Tu. In 1990 the purpose of this extra part was unknown.
Zinoni assumed that some mRNAs, such as those transcribed from the fdhF gene, contains a structure whose function is to facilitate the binding of tRNASec to UGA. He believed that this structure might be one or more stem-loops. (A stem-loop is a region in RNA where two sequences base pairs to each other and form a double-helix called a stem, while a third sequence, called a loop, connects the two base paired sequences.) The mRNA containing the fdhF protein-message was therefore analyzed for potential stem-loop near the UGA codon, and three possible structures were identified (see Zinoni's figure 1).
First experiment: method
Zinoni created nine fdhF deletion-mutants where random lengths of the fdhF gene's 3'-end was removed. This in order to determine how much of the 3'-end that may be removed before translation of UGA is reduced, and how much that may be removed before translation of UGA ceases completely. Information from such an experiment can be used to assess if any of the three identified mRNA stem-loops are really involved in the translation of the UGA codon.
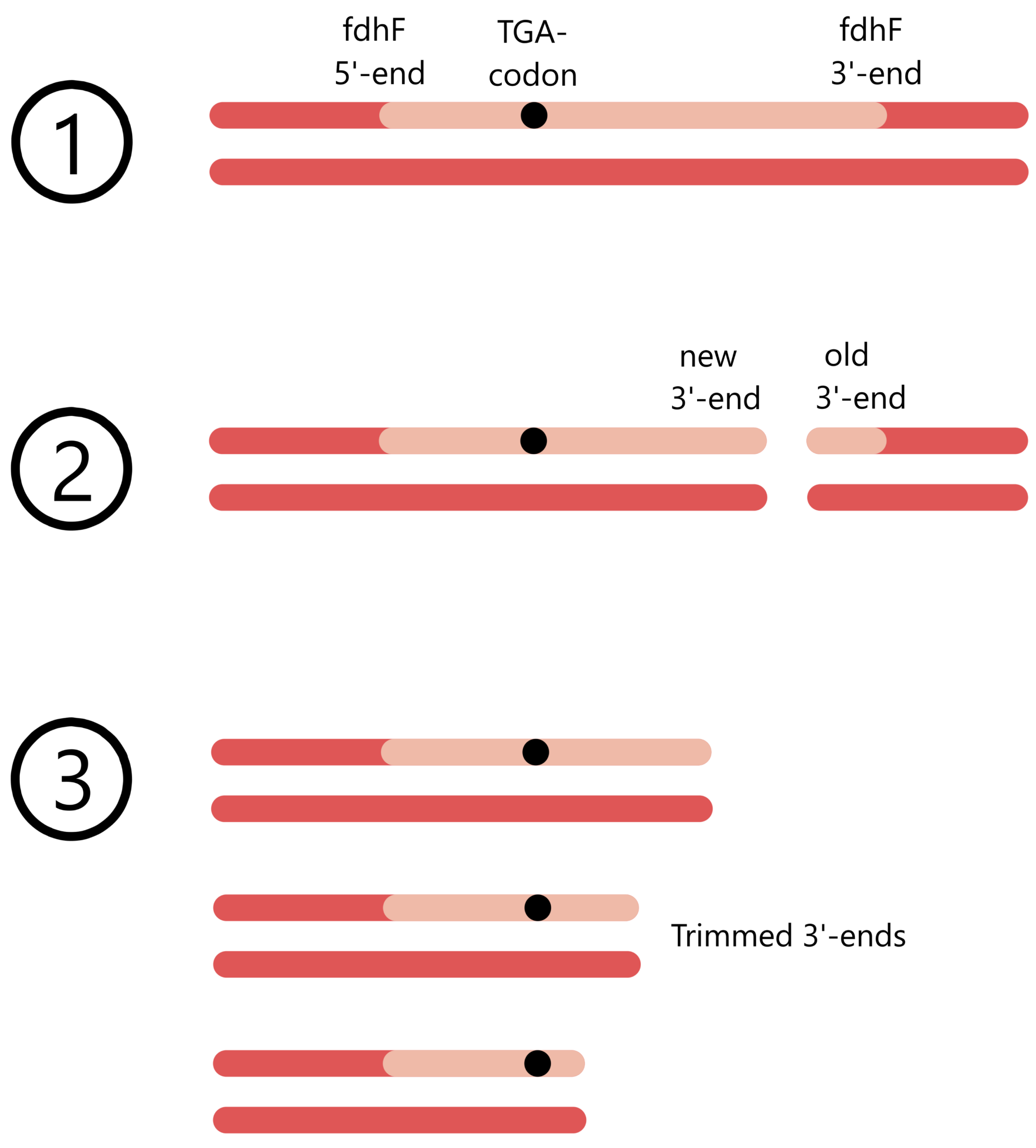
In one mutant, the UGA codon was followed downstream by four bases, while the rest of the 3'-end was removed. This mutant will be referred to as Mut-4. In the other mutants the UGA was followed downstream by 7, 18, 27, 39, 46, 47, 75, and 384 bases, respectively. These will be referred to as Mut-7, Mut-18, etc. (Just for information, the complete fdhF gene contains 1728 bases downstream of the UGA.)
The efficiency of UGA translation in the fdhF deletion-mutants cannot be measured directly, but can be measured indirectly by creating fdhF-lacZ fusion genes, insert the fusion genes into E. coli-cells, and measure something called the β-galactosidase activity.
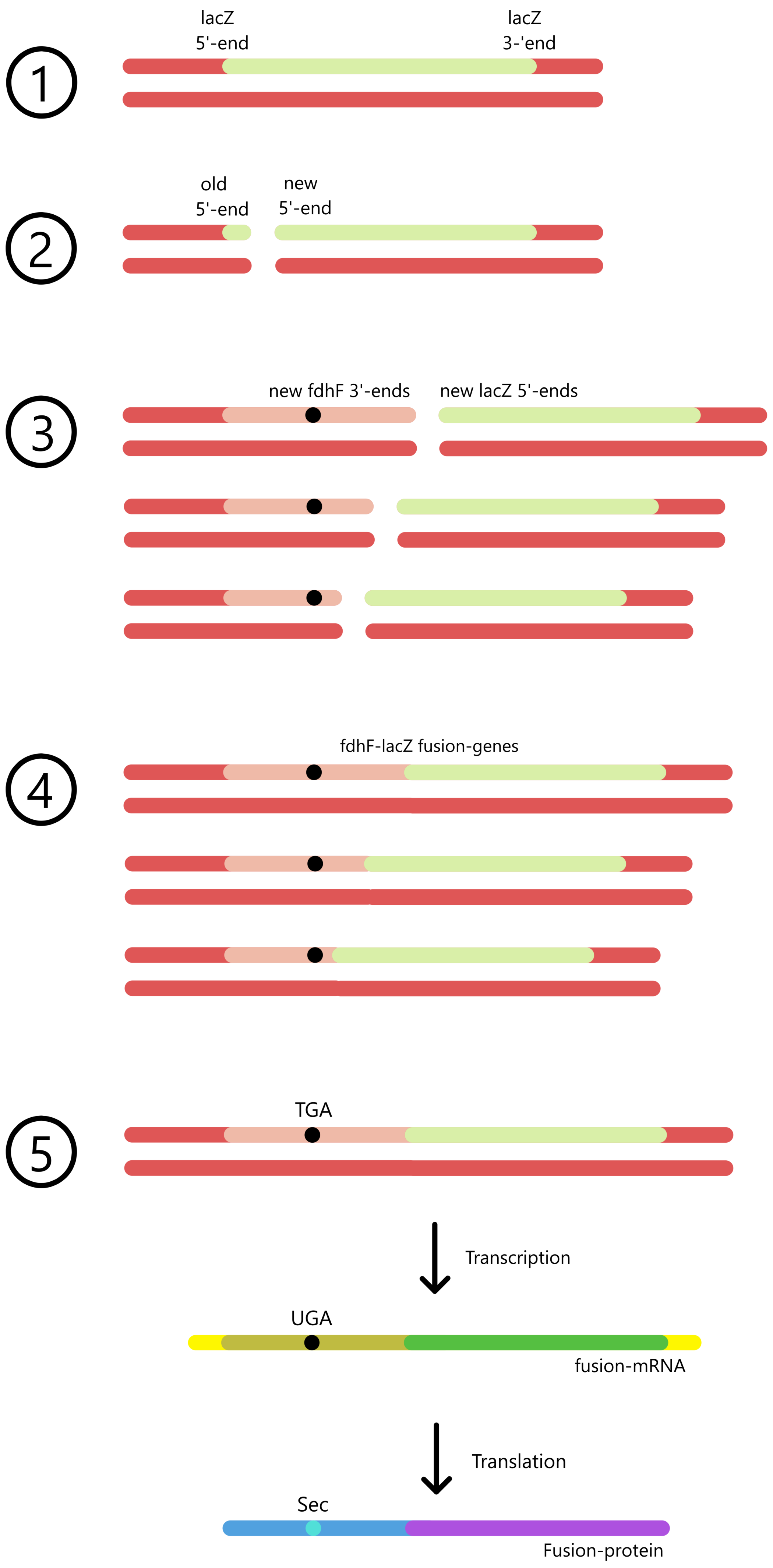
A fdhF-lacZ fusion gene is created by inserting a fdhF mutant near the start (5'-end) of a lacZ gene. E. coli can then transcribe the fusion gene from the fdhF promoter to the lac terminator, creating a fusion mRNA. These mRNA can be translated from the fdhF start codon to the lacZ stop codon, creating a fusion protein. Each of the nine fdhF mutants are inserted into each their fusion gene, which are inserted into each their E. coli culture, so that the β-galactosidase activity can be measured separately for each mutant.
(The lacZ gene in the E. coli chromosome is followed downstream by the lacY- and lacZ genes, where the lac terminator follows behind the lacA gene. This experiment uses a lacZ gene in a plasmid, where lacZ is followed immediately downstream by the lac terminator without other genes in between the two.)
As fdhF was inserted into the 5'-end of lacZ, and since protein messages are translated from the 5'-end towards the 3'-end, the fdhF-part of the message is translated first and the lacZ-part of the message is translated last. This means that translation of lacZ is dependent on translation of fdhF. If the UGA codon of fdhF binds tRNASecACU and is translated to selenocysteine, then lacZ can be translated too, and a fusion protein is created. Alternatively, the UGA can bind RF2 (Release factor 2) and function as a stop codon, in which case lacZ won't be translated and there will be no fusion protein.
The amount of fusion proteins in an E. coli culture can be quantified indirectly by determining the β-galactosidase activity in the culture. lacZ is the gene for β-galactosidase, a protein that splits β-galactosides (for example lactose, which can be split to galactose and glucose). An fdhF-lacZ fusion protein will also have the ability to split β-galactosides. β-galactosidase activity is a measure of how fast β-galactosides are split and is proportional to the amount of fusion proteins.
The β-galactosidase activity is determined by using a laboratory-developed β-galactoside called ONPG (o-nitrophenyl-galactoside), which can be split to galactose and o-nitrophenol. ONPG is colorless and doesn't absorb light, whereas o-nitrophenol absorbs light with a wavelength of 420 nanometers and therefore has a yellow color.
Simply put, the β-galactosidase activity is determined by adding ONPG to the E. coli cultures, waiting a certain time to let the β-galactosidase split the ONPG, then measuring how much light is absorbed by the cultures. Absorbtion of light is measured with a spectrophotometer set to use light with a wavelength of 420 nm. The spectrophotometer displays the results as absorbance values, which are used to calculate Miller values that represent the β-galactosidase activity in the cultures. (Further down in this article there is a section elaborating how Miller values are calculated.)
In other words: a Miller value is proportional to the concentration of o-nitrophenol in a culture, which is proportional to the concentration of fdhF-lacZ fusion genes, which is proportional to how efficiently the UGA codon is translated to selenocysteine.
First experiment: results
The calculated Miller values for the nine different fdhF mutants are presented in Zinoni's figure 2A. Mut-4, Mut-7, and Mut-18 show no UGA translation (Miller values ≈ 0), while Mut-27 and Mut-39 show ineffective UGA translation (Miller values ≈ 200 and 700). Mut-46 has a Miller value of ≈ 1500, this value is maintained in Mut-47 and Mut-75.
This result suggests that Mut-46/47/75 can form the complete mRNA structure necessary for maximal UGA translation, while Mut-27/39 can form incomplete/unstable structures which only facilitate inefficient UGA translation.
In Mut-384 the Miller value increases to ≈ 2200. Considering that the Miller value was constant ≈ 1500 from Mut-46 to Mut-75, Zinoni assumes that the difference in Miller value between Mut-46/47/75 and Mut-384 is caused by "translational reinitiation events", i.e. that Mut-384 don't actually have a more efficient UGA translation than Mut-46/47/75 (I don't know how "translational reinitiation" work, so I cannot make any comment on this).
Of the three mRNA structures presented in Zinoni's figure 1, it is structure 1C that is best supported by this result. Mut-39, which shows reduced UGA translation, cannot form the lowest AU-pair of the 1C stem, while Mut-46, which shows maximal UGA translation, can form all the base pairs of the 1C stem. (I still think it is weird that just one single AU-pair in the stem can cause more than a doubling of UGA translational efficiency, from 700 Miller in Mut-39 to 1500 Miller in Mut-46.)
(If one looks at Zinoni's figure 2A there is a mutant "−7" with a Miller value of ≈ 6000, much higher than in the other mutants. This mutant is not mentioned in the text, but it must be a tenth mutant where the UGA codon was deleted from fdhF together with 7 bases upstream of UGA (and all bases downstream of UGA). The fact that the Miller value increases so much when the UGA codon is removed indicates that translation of UGA is generally an inefficient process relative to the translation of other codons.)
Second experiment
To verify the results of the last experiment, a new experiment was performed using an alternative method. E. coli with the nine mutants were cultured in a medium containing radioactive selenium, so that the bacteria will produce radioactive selenocysteine. Then, when the bacteria produce proteins containing selenocysteine, these proteins will also be radioactive. If the bacteria produce radioactive fdhF-lacZ fusion proteins, this means that the fusion mRNA can form the structure necessary for UGA translation.
Proteins from E. coli were extracted, separated from each other using gel electrophoresis, and analyzed with autoradiography (which makes it possible to see radioactive material). E. coli containing Mut-4/7/18 had no radioactive fusion proteins. Those with Mut-28 had a small amount, those with Mut-39 had a moderate amount, and those with Mut-46/47/75/384 had a large amount (see Zinoni's figure 3). This result is compatible with the Miller values from the first experiment.
Last experiment
In a last experiment, Zinoni created five new fdhF deletion-mutants where varying lengths of the fdhF 5'-end had been deleted. This in order to determine how much of the 5'-end may be removed before UGA translation is reduced. In one mutant, the UGA codon was preceeded by one single base while the rest of the 5'-end was deleted, this will be referred to as Mut-1(5'). In the other mutants the UGA was preceeded by 9, 18, 42 and 67 bases, respectively. These will be referred to as Mut-9(5'), Mut-18(5'), etc.
The mutants were used to create a new set of fdhF-lacZ fusion genes. As in the last experiments, these genes were inserted into E. coli cells, and β-galactosidase activity as well as the amount of radioactive proteins were tested. The result is presented in Zinoni's table 2 and figure 4, and we can see that UGA is being translated in all five mutants. Mut-1(5') has the lowest β-galactosidase activity with 1758 Miller (in E. coli cultured in glycerol), while Mut-9(5') has a significantly higher value of 4908 Miller. In Mut-18(5') and Mut-42(5') the values go a bit down, to 3919 and 4406 Miller, and in Mut-67(5') it goes up to 6144 Miller.
The fact that the β-galactosidase activity increases so much from Mut-1(5') to Mut-9(5') suggests that the first nine bases upstream of the UGA codon are involved in UGA translation. This is compatible with mRNA structures 1A and 1B, but not with structure 1C where the stem-loop doesn't involve any bases upstream of the UGA. It seems strange that the β-galactosidase activity is reduced in Mut-18(5') and Mut-42(5') compared to Mut-9(5'). There is no explanation for this. This reduction is not compatible with mRNA structure 1A, where 18 bases upstream of UGA are needed to form one of the two stem-loops, while 40 bases upstream of UGA are needed to form the entire 1A structure (both stem-loops).
The result from the first experiment fits best with mRNA structure 1C, while the result from the last experiment fits best with mRNA structure 1B. Zinoni proposes a possible mechanism where the mRNA first assumes structure 1B, and when the ribosome approaches the UGA codon and breaks the base pairs upstream of UGA the mRNA assumes structure 1C. With such a mechanism, both structures will be involved in UGA translation.
(The Miller values from the last experiment are generally much higher than those of the first experiment. I think this is because the first experiment uses the fdhF promoter and the fdhF start codon for transcription and translation of the fusion genes, while in the last experiment the lac promoter and lacZ start codon are used. Different promoters and start codons can give different rates of transcription and translation, which will influence the concentration of the fusion proteins. The Miller values from the two experiments should therefore not be compared against each other.)
(Further down in this article there is a section that elaborates on Zinoni's research methods.)
Heider, Bock (1992-a): Targeted Insertion of Selenocysteine into the α Subunit of Formate Dehydrogenase from Methanobacterium formicicum
The bacterium Methanobacterium formicium contains a gene called fdhA. This gene does not contain any selenocysteine codons, and is therefore not translated to a protein containing selenocysteine. Heider attempted to mutate the fdhA gene so as to give it a UGA codon that can be translated to selenocysteine. This was done by altering one of the gene's UGC codons to a UGA codon, and by introducting mutations in the sequence near this UGA as to facilitate the formation of stem-loops similar to those in Zinoni 1990 figure 1.
The mutant fdhA genes were inserted into E. coli cells, and UGA translation was tested with the radioactive selenium method (same method as used by Zinoni). By observing which mutations permit UGA translation and which mutations do not, one can gain a better understanding of the mRNA structure necessary for UGA translation.
Heider first created two mutants called U2 and D2. Mutant U2 can form a stem-loop where the UGA is positioned within the loop. This stem-loop resembles the one presented in Zinoni 1990 figure 1A and 1B (see Heider's figure 1, the stem-loop labelled "E. coli" is the long stem-loop from Zinoni's figure 1A and can also form the short stem-loop from Zinoni's figure 1B). Mutant D2 can form a stem-loop similar to the one in Zinoni's figure 1C (see Heider's figure 2, the stem-loop labelled "E. coli" is the stem-loop from Zinoni's figure 1C).
In Heider's figure 5 we see that there is no UGA translation in either of the two mutants (proteins extracted from mutant U2 are in gel column 6 and proteins extracted from mutant D2 are in gel column 8, neither of the columns contain any protein with radioactive selenocysteine).
The loop in fdhA mutant D2 is quite different from the loop in E. coli's fdhF (see Heider's figure 2). In addition, the stem-loop in E. coli has an uracil protruding from the apical part of the stem, this protruding uracil is absent in mutant D2. Heider therefore created a new fdhA mutant D22 where the apical part of the stem-loop in mutant D2 was replaced by the corresponding part of the E. coli stem-loop (see Heider's figure 2).
In Heider's figure 5 we see that fdhA mutant D22 facilitates translation of UGA (proteins from mutant D22 are in column 14, at the position labelled 93 kDa. I don't know what the weak band below 80 kDa represents.)
The mutations of U2 and D22 were combined into a new mutant called U2D22. This in order to test if the combined U2- and D22-mutations result in a more efficient UGA translation compared to the D22 mutation alone. This was not the case: U2D22 shows a similar amount of UGA translation (similar amount of radioactive protein) as mutant D22 (see Heider's figure 5, proteins from mutant U2D22 are in column 15).
In Zinoni 1990 it was proposed that the stem-loops in 1B and 1C (from Zinoni's figure 1) function together in translation of UGA, in a mechanism where the mRNA first assumes stem-loop 1B, and then switches to stem-loop 1C as the ribosome approaches the UGA codon. Heider's result shows that fdhA mutant D22 facilitates translation of UGA, which supports the idea that stem-loop 1C is involved in the translation of UGA. However, Heider's results does not provide any support to the idea that stem-loop 1B participates in the translation: fdhA mutant U2 had no UGA translation, and the combined mutant U2D22 did not show visibly higher translation of UGA than did mutant D22.
Heider, Baron, Bock (1992-b): Coding from a distance: dissection of the mRNA determinants required for the incorporation of selenocysteine into protein
At this point, it has not been proved that the stem-loop structure in Zinoni 1990 figure 1C is responsible for facilitating translation of UGA, but this is the hypothesis that is best supported by the results of Zinoni and Heider. From here onwards, the base sequence that forms the stem-loop in Zinoni's figure 1C will be referred to as "SECIS" (selenocysteine insertion sequence), and the stem-loop structure will be referred to as the "SECIS-stem-loop".
To investigate how translation of UGA is influenced by mutations in the SECIS-loop, Heider created sixteen fdhF mutants. In each mutant, one of the six bases of the loop was changed to a different base (see Heider's figure 3). UGA translation in the sixteen mutants were tested by creating fdhF-lacZ fusion genes and measuring the β-galactosidase activity of the fusion proteins, and also by checking the amount of radioactive fusion proteins. A fusion gene containing a wildtype (un-mutated) SECIS was used as a reference for effective UGA translation.
The result from the β-galactosidase activity test was that in most of the mutants, UGA translation only measured a few percents relative to the wildtype reference (WT) (see Heider's figure 4A). The test of radioactive fusion proteins showed the same thing: the mutants have little to no radioactive fusion protein compared to the wildtype (Heider's figure 4B, the fusion proteins are at the position labellen "120 kDa"). In other words, it appears that all the bases of the SECIS-loop are important for translation of UGA.
(The two hypothetical loop-variants AGAUCU and AGGCCU were not tested. These two sequences form so-called "reverse complements" and I assume that Heider doesn't test them for this reason, thought he does not comment on it. Personally I would have liked to test those sequences along with the other sixteen mutants, just to have tested every single base change possible.)
In a different experiment, the importance of the length of the SECIS-stem was assessed. Two mutants were created: one with three extra base pairs inserted into the basal part of the stem, and one with three base pairs removed from the basal part of the stem (see Heider's figure 6B). (Note that inserting or removing base pairs like this does not affect the translation of codons, because the reading frame is unaffected when three bases are added or removed. The concept of reading frame is explained in the section elaborating on Zinoni's research methods.)
The mutant with three extra base pairs had a 50% reduced β-galactosidase activity compared to the wildtype reference, while the mutant with three less base pairs had a 100% reduction (see Heider's figure 6C). It therefore appears that the length of the SECIS-stem represents both a minimum-length and an optimal length.
As written in the section on Zinoni 1990: The binding of tRNASecACU to an UGA codon depends on a protein called SelB. This in contrast to every other elongator-tRNA, which depends on the protein EF-Tu for binding to their codons. SelB and EF-Tu have a large similarity in their amino acid sequences, but SelB contains an "extra part" (extra amino acid sequence) at one end of the chain that is not present in EF-Tu.
Heider proposes that the extra amino acid sequence in SelB forms a structure that can bind to the six bases of the SECIS-loop, and that the function of the SECIS-stem is to hold the loop (and thereby SelB + tRNASecACU) in the correct position relative to the ribosome (see Heider's figure 9). This means that when the UGA codon enters the ribosomal A-site, the tRNASecACU will be positioned close to the A-site and can thus bind to UGA before RF2 (release factor 2) has the opportunity to bind. UGA will then function as a selenocysteine codon rather than a stop codon.
(Further down in this article there is a section that elaborates on Heider's research methods.)
Baron, Heider, Bock (1993): Interaction of translation factor SELB with the formate dehydrogenase H selenopolypeptide mRNA
[In vivo-experiments vs in vitro-experiments: Typically one wants to determine how molecules interact inside the cell (that is, in their natural molecular and chemical environment). Experiments which study this are called in vivo (in life) experiments. All research presented in this article are based on in vivo experiments, with exception of Baron 1993, the focus of this section. Certain analyses cannot be performed on molecules inside cells, and for this reason the experiments in Baron 1993 utilize cell-free methods in which a few select molecules are isolated and studied outside of the cell. Such experiments are called in vitro (in glass) experiments. ("Glass" here refers to laboratory glass vials and bottles that contains the molecular solutions. It's not really an ideal name, since in in vivo experiments the cells will be kept in glass containers as well.) The molecular environment can influence how molecules interact together, so one must keep in mind that the results achieved from in vitro experiments are not always representative of how molecules interact inside the cell.]
Heider proposed that SelB, in addition to binding to tRNASecACU and the ribosome, also could bind to the SECIS-loop in the fdhF message. Baron investigated if SelB can bind to an RNA containing the SECIS ("SECIS-RNA") by using a method called "electrophoretic mobility shift assay". This method is based on gel electrophoresis, that is, use of an electric field to drive molecules through a gel. The movement speed of molecules depends on their size, shape, and electric charge. If two molecules bind together, for example a protein and a SECIS-RNA, then this can change their speed through the gel. Such a change is referred to as a "mobility shift", hence the name of the method.
Different concentrations of SelB were added to samples of SECIS-RNA. The SECIS-RNA was radioactive, so as to permit its position in a gel to be determined by autoradiograhpy. The result was that addition of large concentrations of SelB ("10x/25x molar excess") caused a mobility shift (see Baron's figure 1A). This indicates a binding between SECIS-RNA and SelB. With the highest concentration of SelB (25x molar excess) there was, however, a large amount of SECIS-RNA which did not move through the gel at all, and simply remained at the start position.
As mentioned before, the amino acid sequence of SelB is similar to that of EF-Tu, and it was known that EF-Tu must bind GTP (guanosine triphosphate) in order to function in the translation process. Therefore, an additional experiment was performed where SECIS-RNA and SelB were mixed with GTP. A third experiment was also conducted where ATP (adenosine triphosphate) was added to the mixture of SECIS-RNA and SelB. (The structure of ATP is similar to GTP, so Baron wanted to see if ATP could substitute GTP.)
Addition of ATP did not significantly alter the speed of SECIS-RNA through the gel. Addition of GTP, however, caused all the SECIS-RNA to move away from the start position (see Baron's figure 1A), and Baron comments that GTP increases the gel solubility of the RNA/SelB complex. (I assume this means that SelB, in the absence of GTP, has a tendency to bind together with other SelB and create clumps, and that the clumps are too large to move away from the start position, so that SECIS-RNA bound to the clumped SelB therefore remains at the start position.) This doesn't prove that SelB depends on GTP-binding to function in UGA translation, but it does show that GTP has an interaction with the RNA/SelB complex.
Binding of a protein, such as SelB, to an RNA can be either specific (binding to a specific RNA sequence or structure) or non-specific (binding to any RNA sequence). To test if SelB binds specifically to RNA containing the SECIS, samples of SelB were mixed with SECIS-RNA and various other RNAs, such as 5S rRNA. If SelB can bind to any RNA sequence, then some of the SelB proteins will bind to the other RNAs, so that less SelB is available for binding to SECIS-RNA. This means that a smaller amount of SECIS-RNA should be mobility shifted.
The result of these samples is not displayed in a figure, but Baron comments that there was no reduction in the mobility shift of SECIS-RNA. This indicates that SelB binds specifically to the SECIS-part of RNA.
The mobility shift assay was also used to assess whether or not SelB can bind to a SECIS-RNA where the third base of the SECIS-loop has been mutated from a guanine to a cytosine. Heider 1992 showed that this mutation reduced translation of UGA by 99% compared to his wildtype reference (see Heider's figure 4A, in the column labelled C3).
Baron found that the binding between SelB and the mutant SECIS-RNA was significantly weaker that the binding between SelB and un-mutated SECIS-RNA (see Baron's figure 1B). This suggests that the guanine on the third position of the SECIS-loop is involved in the binding between SECIS-RNA and SelB. The hypothesis that SelB binds to the loop of the SECIS structure is therefore supported.
Klug, Huttenhofer, Kromayer, Famulok (1997): In vitro and in vivo characterization of novel mRNA motifs that bind special elongation factor SelB
Heider proposed a model for translation of UGA where SelB binds to the SECIS-loop while the SECIS-stem holds the loop (and thus SelB + tRNASecACU) in the correct position in relation to the ribosome (see Heider 1992-b figure 9). To challenge this model, Klug attempted to find mutant SECIS stem-loops which both can bind to SelB and which has the correct length of the stem (a length similar to the un-mutated SECIS-stem), but still CANNOT facilitate translation of UGA.
In contrast to the previous research presented in this article, where a small number of specific mutations were created, Klug instead created a large number of SECIS-mutants with random mutations. In total there were 5 × 1014 different mutants created, where the average mutant had twelve randomly mutated bases. (The entire SECIS has 39 bases, and Klug uses a random mutation rate of 30% per base, which means that the average mutant will have 0.3 × 39 ≈ 12 mutated bases, but many mutants will have more or less than twelve seeing as the process is random.)
SelB and SEICS mutants were poured over a filter, the pores of which had a diameter of 0.45 micrometer. SelB is too large to pass through the pores, so that SECIS-mutants bound to SelB were retained in the filter, while unbound SECIS mutants passed through. To determine the sequences of the SelB-binding mutants, the RNA from the filter was reverse transcribed to DNA before it was DNA-sequenced.
The sequencing identified thirty different SECIS-mutants able to bind SelB. Nineteen of the mutants contained an "invariant region" in the apical part of the stem-loop (from base G16 to C30), where the bases in these mutants were identical to the un-mutated wildtype SECIS (see Klug's figure 1B, the SECIS wildtype (wt fdhF) is displayed at the top while the different mutants are referred to as "clones").
The remaining eleven mutants had greater sequence variation in the apical part of the stem-loop relative to the wildtype SECIS (see Klug's figure 1C). Only twelve of the thirty mutants were used in further experiments: one mutant with an invariant region, and all eleven mutants with greater sequence variation.
Further experiments
The binding strength between SelB and the twelve select SECIS mutants were investigated. Each SECIS mutant was mixed together with the SECIS wildtype and with SelB, with the concentrations of the three being 10 nanomolar SelB, 40 nanomolar mutant, and 200 nanomolar wildtype. This means that in each sample the mutant and the wildtype have to "compete" for binding to a limited amount of SelB. (I don't know why Klug chose to use five times as much wildtype as mutant, it seems more logical to me to use an equal amount of the two). Each sample was poured over a 0.45 micrometer filter.
To determine the binding ratio of the two RNAs retained in the filter (i.e. the number of SelB-bound mutants per SelB-bound wildtype), the mutants and wildtypes were first separated from each other by gel electrophoresis (this was possible because the two RNAs had different lengths). Before the two RNAs had been mixed together with SelB, both RNAs had been made radioactive by attaching a radioactive phosphate to the 5'-end of each individual RNA molecule. The relative amount of mutant and wildtype in each sample could therefore be determined based on the radioactivity of the two gel bands containing the two RNAs.
The binding ratio of each of the twelve mutants are presented in Klug's table 1. Mutant #945, the one that contained an invariant region, had a binding ratio of 2.0 (twenty SelB-bound mutant RNAs per ten SelB-bound wildtype RNAs in the filter). The eleven other mutants had binding ratios between 0.1 and 3.0. This means that there are some SECIS mutants that bind to SelB better than the SECIS wildtype.
In a last experiment, four SelB-binding SECIS mutants, #945, #922, #934, and #979, were tested for ability to facilitate translation of UGA (the binding ratioes were 2.0, 0.5, 0.8, and 0.7, respectively). Heider 1992-b showed that the length of the SECIS stem is important for UGA translation, and Klug comments that the four chosen mutants can form stems of roughly the same length as the wildtype (see Klug's figure 4A). Translation of UGA was measured indirectly through β-galactosidase activity as in Zinoni 1990.
The calculated Miller values for the four SECIS mutants as well as the SECIS wildtype are shown in Klug's table 2. We see that mutant #945 (the one of the four mutant to have an invariant region) has a value of 970, while the wildtype (WT 60.2) has a value of 1100. Mutant #945 can therefore facilitate UGA translation quite well. In the other mutants #922.1, #934.1, and #979.1 the Miller values are close to zero, so these mutants cannot facilitate UGA translation. This shows that binding of SelB to a SECIS stem-loop with the correct stem length is in itself not enough to facilitate the translation of UGA.
An updated model
As mentioned in the start of this section, Heider proposed a model for translation of UGA where SelB binds to the SECIS-loop, while the SECIS-stem holds the loop in the correct position in relation to the ribosome.
Based on his result, Klug suggests an update of this model. He proposes that the SelB protein may exist in an active or an inactive form, where the inactive SelB can bind to tRNASec and to the SECIS-loop, but cannot translate UGA. Only the active form of SelB can bind to the ribosome and permit translation of UGA.
In the updated model, the ability of the SECIS stem-loop to bind to SelB and hold it in the correct position in relation to the ribosome's A-site is not enough to permit UGA translation. In addition, the SECIS stem-loop must have a structure that induces activation of the bound SelB. Such a mechanism might be economical for the cell, as it can prevent the SelB-tRNASec complex from incorrectly binding to UGA codons supposed to function as stop codons (in which cases there will be no SECIS stem-loop to activate SelB).
Liu, Reches, Groisman, Engelberg-Kulka (1998): The nature of the minimal “selenocysteine insertion sequence” (SECIS) in Escherichia coli
The SECIS stem-loop in E. coli's fdhF gene contains fourteen base pairs (including a possible GU pair) from A3-U42 at the base of the stem to C20-G27 at the apex of the stem (the bases are numbered from the first base of the UGA codon, see Liu's figure 1A where the SECIS wildtype is labelled "a").
Baron 1993 showed that SelB can bind to RNA containing the SECIS stem-loop. Later, Kromayer 1996 showed that SelB's domain 4b could bind to a mini-SECIS containing the full loop and a mini-stem of five base pairs, from G15-C31 to C20-G27. (Kromayer did not test whether or not the mini-SECIS could facilitate translation of UGA.)
The results in Zinoni 1990 indicated that effective translation of UGA depended on all fourteen base pairs in the SECIS-stem. In light of Kromayer's results, Liu wanted to re-examine if all fourteen base pairs really were required for translation of UGA, or if a SECIS-stem with fewer base pairs could also facilitate effective translation. To create stems of reduced length, specific bases in one half of SECIS were mutated so as to be identical to the opposite base in the other half of the stem. For example, a CG-pair can be mutated to G and G. This prevents specific base pairs from forming.
In SECIS-mutant 1b, four base pairs from C4-G41 to G8-U38 were prevented. This caused a ≈ 20% reduction of β-galactosidase activity compared to the un-mutated SECIS wildtype (see Liu's figure 1). In mutant 1c, eight base pairs from C4-G41 to U13-A33 were prevented, with the same ≈ 20% reduction of β-galactosidase activity. In mutant 1d, nine base pairs were prevented, where the ninth pair was a G15-C31 pair that was changed to C15 and C31. This resulted in over 90% reduction of β-galactosidase activity. This result therefore demonstrates that translation of UGA can function fairly well (but not optimally) with mutant 1c's mini-SECIS containing only five base pairs from G15-C31 to C20-G27.
Regarding mutant 1d, it was thought that the reduction of UGA translation was caused either by the lack of base pairing between the bases at positions 15 and 31, or alternatively, was caused by the lack of a guanine at position 15. Another mutant 1e was therefore created, in which the wildtype G15-C31 pair was "flipped" to a C15-G31 pair. In this mutant the bases at positions 15 and 31 can still base pair, while at the same time there is no guanine at position 15. In this mutant the β-galactosidase activity was only reduced 20% compared to the SECIS wildtype, the same reduction seen in mutants 1b and 1c (see Liu's figure 1). A guanine at position 15 is therefore not a requirement.
Based on mutant 1c with the mini-stem of five base pairs, four new mutants were derived to test how the distance between the UGA codon and the mini-stem influences UGA-translation. One mutant had three bases deleted between UGA and the mini-stem, another mutant had six bases deleted, a third mutant had three bases inserted between UGA and the mini-stem, and a fourth had six bases inserted (see Liu's figure 2).
It was found that both deleting and inserting three bases caused a ≈ 50% reduction of β-galactosidase activity compared to the SECIS wildtype, while deleting or inserting six bases caused >90% reduction in β-galactosidase activity. It is therefore clear that the distance between UGA and the mini-stem is of importance in translation of UGA, and it seems like the wildtype-length (eleven bases) is optimal.
The bulging uracil
The SECIS stem contains two uracils (U17 and U18) positioned opposite of one single adenine (A29). This means that one of the two bases cannot partake in a base pair and has to protrude out to the side of the stem. That question is then: is a protruding base at this part of the stem required for translation of UGA? And if so, does the protruding base necessarily have to be at position 17 or 18, or can both positions be used? And does the protruding base have to be a uracil, or can an adenine, cytosine, or guanine also do the job?
Four SECIS-mutants were created in order to answer the first questions. In mutant 3b, the U18 was changed to A18, so that a possible base pair between positions 18 and 31 are prevented (but U17 and A31 can still base pair). This mutant will have a protruding adenine at position 18. In mutant 3d the U17 was changed to A17, so that a possible base pair between position 17 and 31 are prevented (but U18 and A31 can still base pair). This mutant will have a protruding adenine at position 17. Mutants 3b and 3d both had a >90% reduction of β-galactosidase activity compared to the wildtype (see Liu's figure 3).
In mutant 3c the possible U18-A31 pair in the SECIS wildtype was flipped to a A18-U31 pair, so that there will be a protruding uracil at position 17. And in mutant 3e the possible U17-A31 in the wildtype was flipped to a A17-U31 pair, so that there will be a protruding uracil at position 18. Mutant 1c with a protruding uracil at position 17 showed the same high β-galactosidase activity as the wildtype, while mutant 3e with a protruding uracil at position 18 had a >90% reduction in β-galactosidase activity, similar to mutants 3b and 3d (see Liu's figure 3).
Translation of UGA can therefore be facilitated when the stem has a protruding uracil at position 17, but not when the stem has a protruding adenine at position 17, nor with a protruding uracil or adenine at position 18.
Two additional mutants were created, where U17 was changed to C17 and G17, respectively. This to test if a protruding cytosine or guanine at position 17 may be permitted. Both these mutants showed the same >90% reduction of β-galactosidase activity as the mutant where U17 was changed to A17, so the protruding base at position 17 must necessarily be a uracil.
Results that don't agree
Liu's results show that eight base pairs in the basal part of the SECIS stem may be prevented without more than a 20% reduction of UGA translation. This is in disagreement with Zinoni 1990's result, where it was found that Mut-39, where only a single base pair in the base of the stem was prevented, had a 55% reduction in UGA-translation (compared to Mut-46/47/75).
Of this, I think that there must have been something in Zinoni's experiment that negatively influenced translation of UGA in Mut-39. Perhaps the fusion mRNA could form an alternative stem-loop. If a part of SECIS is involved in an alternative stem-loop, then the mRNA may form either the SECIS stem-loop or the alternative stem-loop, but not both at once. Such an alternative stem-loop should therefore be able to reduce translation of UGA. But this is just speculation.
Also, according to Liu's result, deleting three bases between UGA and the mini SECIS-stem causes a 50% reduction of UGA-translation. This is sort of contrary to Heider 1992-b, where it was found that removal of three base pairs in the basal part of the SECIS-stem caused nearly 100% reduction in translation of UGA. Here I have no idea what may be the explanation for the seemingly contradictory results.
Sandman, Tardiff, Neely, Noren (2003): Revised Escherichia coli selenocysteine insertion requirements determined by in vivo screening of combinatorial libraries of SECIS variants
To understand Sandman's experiment, it is necessary to know some details of a virus called M13, and to know about a method called lacZ alpha-complementation.
The M13 virus consists of DNA incapsulated in a protein shell (a capsid) built from five different types of proteins called pIII, pVI, pVII, pVIII, and pIX. If an M13 binds to the surface of an E. coli cell, the viral DNA can enter the cell. Capsid protein pIII is essential for the process of entry. Inside the E. coli cell, the viral DNA is replicated, and the viral genes are transcribed to viral mRNA which is translated into viral proteins. Viral DNA and proteins are assembled into new viruses that are released from the E. coli cell to infect other nearby cells. (The details of the M13 virus are from Smeal 2016. Smeal refers to the viral proteins as p3 instead of pIII, p6 instead of pVI, etc. Sandman uses the name pIII, so in this section I use that name as well.)
lacZ is a gene that is translated to β-galactosidase, the protein that splits β-galactosides such as lactose. By means of genetic engineering it is possible to separate this gene into two smaller genes: lacZΔM15 and lacZα (lacZ-alpha). Neither of these genes can be translated into a functional β-galactosidase, but the two smaller proteins translated from lacZΔM15 and lacZα can bind together and form a functional β-galactosidase. This is called alpha-complementation.
Method
Liu 1998 showed that the SECIS-loop and the apical part of the SECIS stem can facilitate effective translation of UGA, even when the eight base pairs in the basal part of the stem are prevented. This means that significant sequence variation should be permittable in the basal part of the stem.
Sandman investigated how much sequence variation is permitted in the apical part of the SECIS stem, given that the basal part of the stem largely is base paired. This was done by creating SECIS-mutants with random mutations in the apical part of the stem, from U13-A33 to G19-C28 (see Sandman's figure 3). (The method used to create random mutations is the same as in Klug 1997).
Each mutant was inserted into M13 DNA, in the beginning of the gene for protein pIII. The M13 virus' ability to produce pIII proteins and infect new E. coli cells will then depend on the virus having a SECIS mutant that facilitates translation of UGA. (In the same way that production of fdhF-lacZ fusion proteins in Zinoni 1990 depended on the fdhF mutant facilitating translation of UGA.)
The genetically modified M13 were inserted into E. coli cells. These infected E. coli were cultured in a nutrient gel together with non-infected E. coli. The fact that the bacteria are cultured in a nutrient gel rather than in a liquid nutrient solution means that the bacteria cannot move around, they remain at the location they were when the gel solidified.
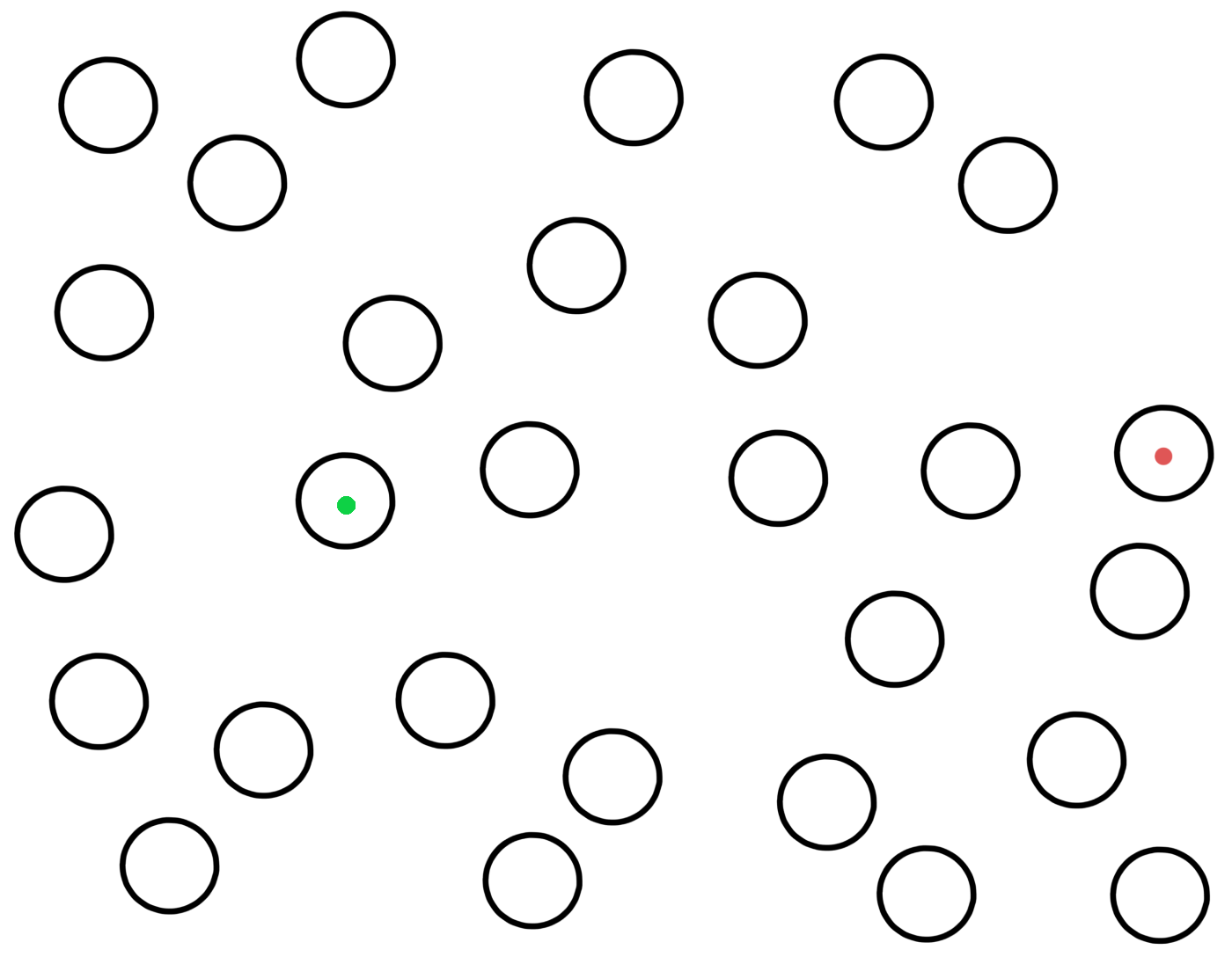
An M13 with a SECIS mutant that facilitates translation of UGA will produce new viruses which are released from the infected E. coli and can infect other nearby E. coli (the viruses are small enough to diffuse through the nutrient gel). These other infected E. coli then produce additional viruses which infect additional nearby E. coli. In this way the infection will spread as a growing circle centered on the original infected E. coli.
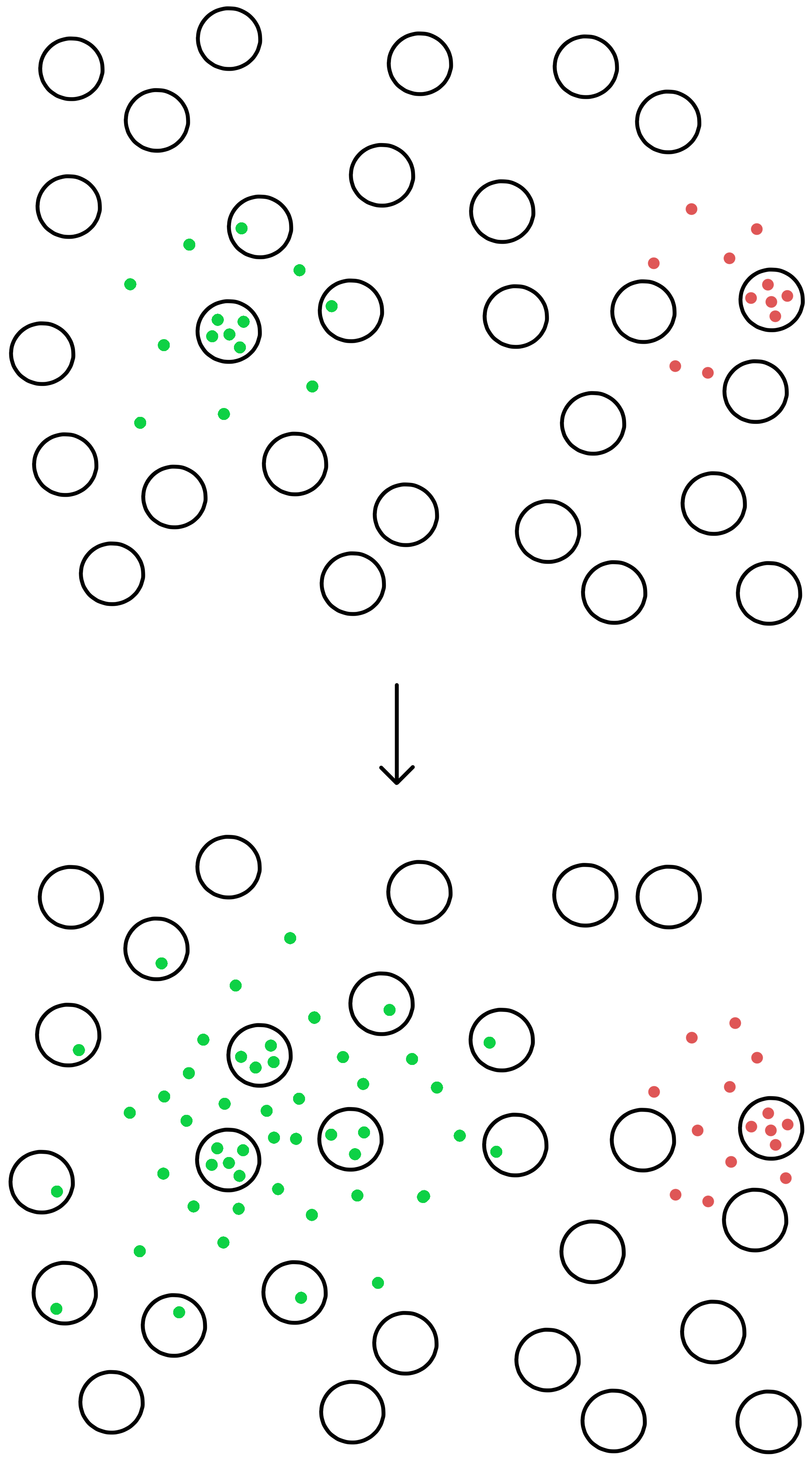
The nutrient gel in which the E. coli cells are cultured contains β-galactosides called X-gal. β-galactosidase can split X-gal to galactose and 5,5’-dibromo-4,4’-dichloro-indigo, where the latter is a blue-colored molecule.
The E. coli cells in the gel do not have a normal lacZ gene, they instead have a lacZΔM15 gene, while DNA from the M13 virus contains a lacZα gene. This means that infected E. coli cells can produce functional β-galactosidases through alpha-complementation, and the β-galactosidases can split X-gal to a blue-colored molecule, so that infected E. coli becomes blue. Uninfected E. coli remain colorless.The infected E. coli can thus be seen as blue dots in the nutrient gel. M13-DNA extracted from these dots is sequenced to identify the SECIS mutants that facilitate translation of UGA.
X-gal vs ONPG
In Sandman's experiment, the activity of β-galactosidase is tested with X-gal, which is split to the blue-colored molecule dibromo-dichloro-indigo, whereas in Zinoni 1990 the activity of β-galactosidase was tested with ONPG, which is split to the yellow-colored molecule o-nitrophenol. There's more than the color that differentiate the two: o-nitrophenol is water soluble, while dibromo-dichloro-indigo is not.
When Sandman uses X-gal to localize infected E. coli cells in the nutrient gel, the dibromo-dichloro-indigo will remain in the area where X-gal was split, so that only areas containing infected E. coli turn blue. The way I understand it, if Sandman had used ONPG instead of X-gal, then the entire nutrient gel would have turned yellow and it would have been impossible to determine the location of the infected cells. This is because o-nitrophenol is water soluble and can therefore diffuse throughout the gel (which is water-based). This doesn't happen with dibromo-dichloro-indigo because this molecule isn't water soluble.
In Zinoni's experiment, on the other hand, it was necessary to use ONPG. In his experiments the β-galactosidase activity of the bacteria was determined by culturing the bacteria in liquid (water-based) nutrient solutions and using a spectrophotometer to measure the absorbance in the solutions. As o-nitrophenol is water-soluble, these molecules will spread evenly throughout the nutrient solutions. This is a prerequisite for using a spectrophotometer to estimate the concentration of o-nitrophenol. If Zinoni had used X-gal instead of ONPG, I believe the dibromo-dichloro-indigo molecules would have clumped together, because as they are not water-soluble. This would have made it impossible to estimate the molecule's concentration with a spectrophotometer.
Results
Sandman identified nineteen different SECIS with mutations in the apical part of the stem (see Sandman's figure 3). The efficiency of UGA translation was tested in a similar manner as in Zinoni 1990 (see Sandman's figure 5).
(A comment to Sandman's figure 5: Red bars represents values from bacteria cultured in a medium with selenium. Blue bars represents control values from bacteria cultured without selenium, where UGA cannot be translated to selenocysteine (UGA can still be translated to tryptophan, although this translation is not very effective, so a small amount of functional β-galactosidases are created even when UGA cannot be translated to selenocysteine). The SECIS wildtype is referred to as "TGACwt". The β-galactosidase activity of bacteria with the SECIS wildtype, cultured with selenium, are used as a reference value. The red bar of the wildtype therefore shows a value of 1 (= 100% β-galactosidase activity). The β-galactosidase activity in the SECIS mutants are given relative to this reference value, so that for example a value of 1.4 would mean that the β-galactosidase activity is 40% higher than in the wildtype.)
Liu 1998 showed that the SECIS's protruding U17 is important in translation of UGA, and that this base cannot be replaced by a protruding U18 nor by a protruding A17/C17/G17. In Sandman's SECIS mutants SV15 and SV17, the wildtype's U17 has been changed to a G17, while a G16 has been changed to a U16. Therefore, a protruding U16 can replace a protruding U17, although with a reduction in translation of UGA (about 50% reduction in mutant SV15 and 85% reduction in SV17, relative to the SECIS wildtype).
Sandman's result also shows that UGA can be translated efficiently even when not all the apical bases of the stem are base paired. Eighteen of the mutants contain one prevented base pair among the five base pairs closest to the loop, and many of these mutants facilitate high UGA-translation. In one mutant, SV13, three of the five base pairs closest to the loop are prevented. This mutant has lower translation of UGA than most other mutants.
Together with the result of Liu 1998, Sandman's result shows that base pairing in the apical part of the SECIS stem is more important than base pairing in the basal part of the stem, but the apical part of the stem doesn't necessarily need to be completely base paired to achieve translation of UGA.
Summary
It would probably have been possible to write much more about SECIS, but I feel that this article now describes SECIS in E. coli quite well, so I will stop here. Here's a short summary of what has been presented:
A UGA codon can function either as a stop codon or as a selenocysteine codon. In order to function as a selenocysteine codon, it is necessary that the UGA be followed downstream by a SECIS (selenocysteine insertion sequence). In E. coli's fdhF gene the SECIS consists of 39 bases just downstream of UGA. The SECIS can form a stem-loop where the six bases of the stem, as well as one protruding uracil in the stem, assumably form the contact points responsible for binding to and activating the SelB protein. The active SelB then allows tRNASec to bind to the UGA codon. The SECIS-stem must have a certain length, not too short and not too long, in order to correctly position SelB relative to UGA. The stem does not need to be fully base paired, especially not the basal part of the stem.
A detailed description of Miller's method for measuring β-galactosidase activity
This section is an appendix to the earlier section on Zinoni 1990.
The “Miller-method” for testing β-galactosidase-activity in a liquid cell culture was originally described in the book “Experiments in Molecular Genetics” (Jeffrey Miller, 1972). The essence of the method can be summarized like this: add ONPG (o-nitrophenyl-galactoside) to a liquid E. coli-culture, wait until a noticable yellow color appears in the culture, and measure the culture’s optical density at a wavelength of 420 nanometer (that is, measure the OD420). Miller used this “activity-formula” to calculate the β-galactosidase-activity:
Activity units (commonly called Miller units) =
1000 × (OD420 – 1.75 × OD550) / (time × volume × OD600)
To understand the formula it is necessary to understand the Miller-method in more detail. First, the E. coli-cells are cultured until they are growing rapidly (until they are in the middle of the exponential part of their growth curve). At this point, the culture vessel is placed into ice water to cool the bacteria and inhibit further growth. 1 mL of the cooled culture is withdrawn to determine the cell concentration by measuring the OD600 in a spectrophotometer. (The cell concentration is proportional to the value of OD600.) The measured OD600 is inserted into the activity-formula.
Another volume of the cooled culture is withdrawn and mixed with a solution called “Z-buffer”. If one expects the bacteria to have a high β-galactosidase-activity, withdraw 0.1 mL of the culture and mix with 0.9 mL of Z-buffer. If one expects the bacteria to have a low β-galactosidase-activity, withdraw 0.5 mL of the culture and mix with 0.5 mL of Z-buffer. The mix of bacteria and Z-buffer will be referred to as the “reaction mix”. The volume of cooled culture used in the reaction mix, either 0.1 or 0.5 mL, is inserted into the activity-formula.
(Miller does not explain the function of the Z-buffer, but according to this protocol the Z-buffer is designed to stabilize the β-galactosidases (to prevent them from becoming inactivated over time). The stabilization is due to one component of the Z-buffer, β-mercapto-enthanol, which functions as an anti-oxidant and protect the β-galactosidases from reaction with oxygen. I assume such a reaction would interfere with the β-galactosidase’s activity.)
A drop of toluene is added to the reaction mix. Miller writes that “The toluene partially disrupts the cell membrane, allowing small molecules such as ONPG to diffuse into the cells.” (So I guess the toluene causes small holes to develop in the membranes.)
Next, the temperature of the reaction mix is set to 28°C, ONPG is added, and the reaction start time is recorded. The β-galactosidases in the E. coli-cells will begin to split ONPG into galactose and o-nitrophenol, where the latter is a yellow-colored molecule. Once the reaction mix has developed a noticable yellow color (which might take minutes or hours), sodium carbonate is added. Sodium carbonate changes the pH of the reaction mix from pH 7 to pH 11, which inactivates the β-galactosidases and prevents further splitting of ONPG, so that the intensity of the yellow color does not increase further. The reaction end time is recorded. The reaction start time and end time is used to calculate the reaction time (in minutes), which is inserted into the activity-formula.
(Why is the reaction performed at 28°C rather that another temperature, for example 37°C? The latter temperature would increase the reaction speed. I am not sure, but I guess that the β-galactosidases are significantly more stable at 28°C than at 37°C, and that Miller chose 28°C for this reason. Also note that even if the temperature is raised to 28°C the E. coli cannot resume growth, because their cell membranes have holes after the toluene-treatment. The cell concentration in the reaction mix will therefore remain constant throughout the reaction.)
At pH 11 the o-nitrophenol absorbs light most efficiently at a wavelength of 420 nm (the absorbance spectrum of o-nitrophenol is shown in Figure 2 in this PDF). The concentration of o-nitrophenol in the reaction mix is proportional to how much light the reaction mix absorbs at 420 nm. Therefore, the OD420 of the reaction mix (optical denisty at 420 nm) is measured in a spectrophotometer.
However, light absorbation by o-nitrophenol is not the only factor that influences the OD420 of the reaction mix. Light scattering by bacterial cell material (or “cell debris” as Miller refers to it) also contributes to the OD420 value. We can therefore think of the OD420 as being the sum of ODabsorbed420 and ODscattered420, where it is the ODabsorbed420 that we really wish to determine.
ODtotal420 = ODabsorbed420 + ODscattered420
When measuring OD at a wave length of 550 nm there will be zero absorbation by o-nitrophenol (which can be seen on the absorbance spectrum linked above). Only light scattering by cell material will contribute to the OD550 value.
ODabsorbed550 = 0
ODtotal550 = ODscattered550
The degree of light scattering by cell material is not the same when measuring OD at 420 and 550 nm wavelengths. According to Miller,
ODscattered420 = 1.75 × ODscattered550
By combining the above equations we get:
ODabsorbed420 = ODtotal420 – 1.75 × ODtotal550
Therefore, OD420 – 1.75 × OD550 is inserted into the activity-formula.
Lastly, the fraction (OD420 – 1.75 × OD550) / (time × volume × OD600) is multiplied by 1000. By doing this, cells with very low β-galactosidase-activity will yield about 1 Miller-unit. (If we don’t multiply by 1000 then cells with very low β-galactosidase-activity will yield about 0.001 Miller-unit. I guess Miller just prefers integers over decimals and that’s why he multiplies by 1000 in the activity-formula.)
The purpose of the term time × volume × OD600: what the activity units represent
We can now consider what the activity formula really means. The expression OD420 – 1.75 × OD550 is proportional to the concentration of o-nitrophenol in the reaction mix at the time when the reaction is ended. The expression (OD420 – 1.75 × OD550) / time is proportional to the increase in o-nitrophenol concentration per minute during the reaction. This increase in concentration is itself proportional to the β-galactosidase-activity in the reaction mix.
The value of OD600 is proportional to the cell concentration (number of cells per volume) in the bacterial culture, so the expression volume × OD600 is proportional to the number of E. coli-cells that are present in the reaction mix. The expression (OD420 – 1.75 × OD550) / (time × volume × OD600) is therefore proportional to the β-galactosidase-activity per E. coli-cell in the reaction mix. This is what the activity-formula calculates.
Example calculation of β-galactosidase-activity
Suppose that we allow a culture of E. coli to grow until it measures an OD600 of 0.562. We expect a high β-galactosidase-activity and therefore use a culture volume of 0.1 mL in the reaction mix. We start the reaction, and every 15 minutes we check the color of the reaction mix. After 75 minutes a noticable yellow color has appeared and the reaction is stopped. We measure an OD420 of 0.833 and an OD550 of 0.102. The β-galactosidase-activity can then be calculated to
1000 × (0.833 – 1.75 × 0.102) / (75 × 0.1 × 0.562) = 155 Miller units
Alternative activity-formula without OD550 measurement
Miller notes that after the reaction is stopped by addition of sodium carbonate, it is possible to centrifuge the reaction mix in order to separate the cell material from the liquid. The cell material will congregate into a little “pellet” at the bottom of the tube containing the reaction mix. By adding this centrifugation-step the ODscattered420 should equal 0, which eliminates the need for the term – 1.75 × OD550. The activity-formula then becomes:
Miller units = 1000 × OD420 / (time × volume × OD600)
(I have the feeling that this alternative version of the method/formula would calculate the β-galactosidase-activity more accurately, and centrifuging the reaction mix shouldn’t take much time, so I don’t know why this isn’t the standard formula.)
The amount of ONPG added to the reaction mix is important
When using the Miller method to determine β-galactosidase-activity, the activity has to be proportional to the number of β-galactosidases per E. coli-cell. For this to be true it is necessary that all β-galactosidases be splitting ONPG continuously. That is, the β-galactosidases must be 100% saturated by ONPG. Therefore, the concentration of ONPG added to the reaction mix has to far exceed the concentration of β-galactosidase present in the reaction mix.
If only a relatively small amount of ONPG is added, then only some of the β-galactosidases will be saturated. The degree of saturation might be 1% (i.e. at any moment only 1% of all the β-galactosidases are in the process of splitting ONPG). Or the saturation might be 90%, or it might be something else. Without knowing the degree of saturation we cannot use the β-galactosidase-activity as an indirect measure for the number of β-galactosidases.
Miller adds 0.2 mL of ONPG solution (4 mg ONPG per mL water) to the 1 mL of reaction mix. With his experimental setup this suffices to achieve 100% saturation of the β-galactosidases.
(If you use the Miller-method to measure β-galactosidase-activity with a different setup, it should be easy to test whether using 4 mg ONPG per mL water gives a sufficiently concentrated ONPG-solution to fully saturate the β-galactosidases. You’d create two reaction mixes, add 0.2 mL of the 4 mg ONPG solution to one reaction mix, then add 0.2 mL of an alternative 6 mg ONPG solution to the second reaction mix. If the 4 mg solution is enough to 100% saturate the β-galactosidases, then the 6 mg solution will also give 100% saturation, and the two reaction mixes should yield the same OD420 value. If the two mixes were to yield different OD420 values then this would show that the 4 mg solution, and perhaps also the 6 mg solution, do not fully saturate the β-galactosidases. You would then have to make more concentrated solutions of ONPG and see if those result in the same OD420.)
An explanation of the research method from Zinoni, Heider, Bock (1990): Features of the formate dehydrogenase mRNA necessary for decoding of the UGA codon as selenocysteine
This section is an appendix to the previous section on Zinoni 1990. I will be quoting from Zinoni's "Materials and methods".
For generation of 3’ deletions of the fdhF gene, plasmid pFM22 was constructed by deletion of a 1.86 kilobase EcoRV/Xba I fragment from plasmid pFM30, thereby introducing a Bgl II site at codon 268 of fdhF.
Plasmid pFM30 contains a copy of the fdhF gene (according to Zinoni's reference 9). In fdhF, codons number 269 and 270 constitute a EcoRV restriction site (to determine this I checked the gene sequence of fdhF in the BioCyc database). 1860 bases downstream of this EcoRV site (downstream along the fdhF non-template strand) there is an Xba I restriction site.
5'...TTGAAGATATCACCGG...(1850 bases)...XXXXXTCTAGAXXXXX...3'
3'...AACTTCTATAGTGGCC...(1850 bases)...XXXXXAGATCTXXXXX...5'
(I don't know the sequence around the Xba I site, so the bases around it are symbolized by "X".)
By use of RcoRV and Xba I restriction nucleases, plasmid pFM30 was cut at the two mentioned restriction sites so that two fragments were created: one fragment (with 1860 bases) contained the 3'-end of the fdhF gene, while another fragment contained the 5'-end of the fdhF gene along with the fdhF promoter.
3'...AACTTCTA TAGTGGCC...(1850 bases)...XXXXXAGATC TXXXXX...5'
The fragment containing the 5'-end of fdhF was isolated with gel electrophoresis. The fragment-end that was cut by EcoRV is a blunt end, while the end that was cut with Xba I is a 5'-overhanging end. The latter is "filled in" and turned into a blund end by using DNA polymerase I (Klenow), so that the fragment has two blunt ends.
3'...AACTTCTA GATCTXXXXX...5'
The fragment ends are ligated together to form a circular DNA. A Bgl II restriction site is formed at the ligation site. This Bgl II restriction site starts at the third base of fdhF codon number 268 and ends at the second base of codon number 270.
5'...TTGAAGATCTAGAXXXXX...3'
3'...AACTTCTAGATCTXXXXX...5'
The result is the creation of a new plasmid pFM22, about 1860 bases shorter than pFM30, where fdhF codon number 268 is followed by a Bgl II restriction site rather than an EcoRV restriction site.
(To be honest, although I understand what is being done here I do not understand why it is being done, why it should be advantageous to create plasmid pFM22 rather than simply doing the next part of the experiment with plasmid pFM30.)
Plasmid pFM22 was linearized with Bgl II and then treated with exonuclease BAL-31 at a degradation rate of 50 base pairs per minute.
The Bgl II restriction site of plasmid pFM22 was cut with a Bgl II restriction nuclease, in order to make the plasmid linear. fdhF's codon 268 is positioned at one end of the plasmid and now constitute the 3'-end of fdhF's non-template strand. The exonuclease BAL-31 is used to cut off bases from the plasmid ends, so that the fdhF non-template strand is cut from its 3'-end towards its 5'-end.
After a given time BAL-31 is inactivated by increasing the temperature, so that the cutting is stopped close to fdhF's UGA codon. By using multiple samples of the plasmid and BAL-31 and varying the time before inactivation of BAL-31, different fdhF 3' deletion mutants are created.
fdhF's UGA codon is codon number 140, while the Bgl II restriction site is positioned 3' of codon 268. It is therefore 128 codons, or 384 bases, that separate the two. Zinoni's mutant Mut-384 is therefore created without using BAL-31 to cut the plasmid ends.
After «fill-in» of 5’ protruding ends with DNA polymerase I (Klenow), ligation with BamHI linkers (dCGCGGATCCGCG), and restriction with EcoRI and BamHI, the fragments were separated on a 1.5% agarose gel and recloned into EcoRI/BamHI-digested plasmid pACYC184. Inserts were sized by restriction analysis and their exact end points were determined by DNA sequencing.
Cutting the ends of the plasmid with BAL-31 can create both blunt ends and overhanging ends. 5' overhanging ends are filled in with DNA polymerase I (Klenow) so that all plasmid ends become blunt ends. Both ends of the plasmid are ligated to synthetic "linker-DNAs", which each contain a BamHI restriction site. ("d" in front of the base sequence of the linker (dCGCGGATCCGCG) means "deoxy" and is used to specify that the linker is a DNA sequence, not an RNA sequence.)
For example, one of the 3' deletion mutants produced by BAL-31 is Mut-7, where the UGA codon is followed downstream by seven bases. Mut-7 is ligated to a linker:
5'...TGACACGGCC CGCGGATCCGCG 3'
3'...ACTGTGCCGG GCGCCTAGGCGC 5'
After ligation:
5'...TGACACGGCCCGCGGATCCGCG 3'
3'...ACTGTGCCGGGCGCCTAGGCGC 5'
(As these sequences are DNA, and DNA contains the base T rather than U, the UGA codon is shown here as TGA.)
After ligation, the plasmid is cut with EcoRI and BamHI restriction nucleases. BamHi cuts the BamHI site in the linker at the fdhF gene's shortened 3'-end, while EcoRI cuts an EcoRI restriction site located somewhere 5' of the fdhF promoter (I'm not sure exactly how far away the restriction site is). The resulting plasmid-fragment, containing the fdhF promoter and the 5'-part of the fdhF gene, is isolated with gel electrophoresis.
The cuts from BamHI and EcoRI has given the fragment two different overhanging ends. A pACYC184-plasmid is also cut by BamHI and EcoRI, so that this plasmid gets the same overhanging ends as the fdhF-fragment. pACYC184 and the fdhF fragment are mixed together, allowing their ends to base pair so they can be ligated together.
To verify that the fdhF fragment was correctly inserted into plasmid pACYC184, the sequences near the ligation sites are subjected to restriction analysis and DNA sequencing.
These fdhF fragments were used to generate lacZ fusions with one of plasmids pFM1400, pFM1401, and pFM1402. They carry the polylinker of plasmid pUC8 in front of a truncated lacZ gene in all three reading frames and differ in only 1 or 2 bases, thus minimizing effects arising from different lacZ linker joints.
The plasmids pFM1400/1401/1402 contain a lacZ gene where the first seven codons have been removed (according to Zinoni's reference 15). (It is possible to remove the first seven codons from lacZ without causing a loss of function of the β-galactosidase protein translated from the gene.) At the 5'-end of lacZ there is a polylinker, a base sequence containing multiple restriction sites clustered together. Here is the sequence of the polylinker in plasmid pFM1401 in front of the lacZ gene (which starts at a codon that corresponds to the eight codon of the wildtype lacZ gene):
/ \ / \ / \ lacZ (non-template)
5'...GAATTCCCGGGGATCCGTCGACCTGCAGCCAAGCTTGCTCTGGCCGTCGTTTTA...3'
3'...CTTAAGGGCCCCTAGGCAGCTGGACGTCGGTTCGAACGAGACCGGCAGCAAAAT...5'
\ / \ / \ /
SmaI AccI HindIII
SalI
Among the polylinker restriction sites is an EcoRI site and a BamHI site, which are used to insert the fdhF fragment in front of lacZ to create a fusion gene. Each plasmid with a fusion gene is inserted into E. coli by means of electroporation.
The fdhF fragments inserted into the plasmids lack a transcriptional terminator, as this has been deleted along with the 3'-part of fdhF. The lacZ gene also lacks a promoter. This means that in the fusion gene the fdhF part and the lacZ part cannot be transcribed individually, they can only be transcribed together as a single mRNA.
The fdhF part contains a start codon, while the lacZ part does not. Translation of the fusion gene may terminate at two points: if the fdhF UGA codon is not translated to selenocysteine (due to the lack of a SECIS structure), then UGA will act as a stop codon, and the resulting protein will only contain a part of the fdhF protein. Alternatively, if UGA is translated to selenocysteine, then translation will stop at the lacZ stop codon. (the fdhF stop codon has been deleted, so translation doesn't stop at the end of fdhF.)
The important point is that lacZ in the plasmid cannot be translated individually, it can only be translated as part of a fdhF-lacZ fusion protein. Plasmids carrying fusion genes are inserted into E. coli where the existing lacZ gene is defective. The fusion proteins will thus be responsible for all β-galactosidase activity in the bacteria, a necessity if the β-galactosidase activity is to be proportional with the concentration of fusion proteins. (As mentioned earlier, this is necessary if the Miller values are to be proportional with the efficiency of UGA translation.)
Why are the fdhF fragments inserted into three different plasmids pFM1400, pFM1401, and pFM1402? All base sequences can be read as three different base triplet sequences, called reading frames. Here is an example from the beginnig of the lacZ gene (non-template strand):
...GAA ACA GCT ATG ACC ATG ATT ACG GAT TCA CTG GCC GTC GTT TTA C...
...G AAA CAG CTA TGA CCA TGA TTA CGG ATT CAC TGG CCG TCG TTT TAC...
...GA AAC AGC TAT GAC CAT GAT TAC GGA TTC ACT GGC CGT CGT TTT AC...
When a gene sequence is translated to a protein, only the reading frame containing the start codon is used:
1st 2nd 3rd 4th 5th 6th 7th 8th 9th 10t 11t 12t...
...GAA ACA GCT ATG ACC ATG ATT ACG GAT TCA CTG GCC GTC GTT TTA C...
Met Thr Met Ile Thr Asp Ser Leu Ala Val Val Leu ...
(corresponding amino acids)
When two genes are fused together, it is necessary that the codons of both genes are in the same reading frame, because both genes are translated from the same start codon.
fdhF and lacZ are connected by a polylinker sequence, and the length of this polylinker determines if the two genes are in the same reading frame or not. Because the experiment includes multiple fdhF mutants with different 3' deletions, it is necessary to use three different polylinkers, each varying in length by one or two bases compared to the other polylinkers (the polylinker sequences are given in Zinoni's reference 15). Each of the three polylinkers are located in each their plasmid (one in pFM1400, one in pFM1401, one in pFM1402). Here is a comparison of the three polylinkers (the variable part of the polylinkers is marked with bold text, the eight lacZ codon (CTG) begins just downstream of the variable part):
5'...GAATTCCCGGGGATCCGTCGACCTGCAGCCAAGCTTGCGATCTGGCCGTCGTTTTA...3'
3'...CTTAAGGGCCCCTAGGCAGCTGGACGTCGGTTCGAACGCTAGACCGGCAGCAAAAT...5'
Polylinker in pFM1401:
5'...GAATTCCCGGGGATCCGTCGACCTGCAGCCAAGCTTGCTCTGGCCGTCGTTTTA...3'
3'...CTTAAGGGCCCCTAGGCAGCTGGACGTCGGTTCGAACGAGACCGGCAGCAAAAT...5'
Polylinker in pFM1402:
5'...GAATTCCCGGGGATCCGTCGACCTGCAGCCAAGCTTCGATCTGGCCGTCGTTTTA...3'
3'...CTTAAGGGCCCCTAGGCAGCTGGACGTCGGTTCGAAGCTAGACCGGCAGCAAAAT...5'
To demonstrate why one must use three different polylinkers: if fdhF mutant Mut-7 is inserted into the polylinker of plasmid pFM1401, then the lacZ codons will be translated into the correct amino acids (compare the lacZ codons and corresponding amino acids which were shown in a previous figure):
fdhF lacZ
5'...TGACACGGCCCGCG GATCCGTCGACCTGCAGCCAAGCTTGCTCTGGCCGTCGTTTTA...3'
3'...ACTGTGCCGGGCGCCTAG GCAGCTGGACGTCGGTTCGAACGAGACCGGCAGCAAAAT...5'
Mut-7 ligated to pFM1401:
fdhF lacZ
5'...TGACACGGCCCGCGGATCCGTCGACCTGCAGCCAAGCTTGCTCTGGCCGTCGTTTTA...3'
3'...ACTGTGCCGGGCGCCTAGGCAGCTGGACGTCGGTTCGAACGAGACCGGCAGCAAAAT...5'
lacZ is translated to the correct amino acids: lacZ-codons
... ... ... ... ... ... ... ... ... ... ... ... ... ... ... 8th 9th 10t 11t 12t ...
5'... TGA CAC GGC CCG CGG ATC CGT CGA CCT GCA GCC AAG CTT GCT CTG GCC GTC GTT TTA ...3'
... Sec His Gly Pro Arg Ile Arg Arg Pro Ala Ala Lys Leu Ala Leu Ala Val Val Leu ...
While if Mut-7 is inserted into to polylinker of plasmid pFM1400 or pFM1402 then lacZ will be translated to the wrong amino acids (marked with red text):
5'... TGA CAC GGC CCG CGG ATC CGT CGA CCT GCA GCC AAG CTT GCG ATC TGG CCG TCG TTT ...3'
... Sec His Gly Pro Arg Ile Arg Arg Pro Ala Ala Lys Leu Ala Ile Trp Pro Ser Phe ...
Mut-7 inserted into pFM1402:
5'... TGA CAC GGC CCG CGG ATC CGT CGA CCT GCA GCC AAG CTT CGA TCT GGC CGT CGT TTT ...3'
... Sec His Gly Pro Arg Ile Arg Arg Pro Ala Ala Lys Leu Arg Ser Gly Arg Arg Phe ...
Mut-18, on the other hand, must be inserted into plasmid pFM1402 if lacZ is to be translated to the correct amino acids, while Mut-47 must be inserted into plasmid pFM1400. The other mutants also have to be inserted into a specific plasmid if lacZ is to be translated correctly.
(Regarding why Zinoni inserts the fdhF fragments into plasmid pACYC184 before the fragments are cut out and inserted into plasmids pFM1400/1401/1402, this is something I don't understand and can't explain.)
An explanation of the research method from Heider, Baron, Bock (1992-b): Coding from a distance: dissection of the mRNA determinants required for the incorporation of selenocysteine into protein
This section is an appendix to the previous section on Heider 1992-b. I will be quoting from Heider's "Materials and methods".
Mutated DNAs [containing the selenocysteine codon and its accompanying selenocysteine insertion sequence] were generated in vitro by annealing either two or four oligonucleotides, filling in single-stranded regions and inserting the products into plasmid pUC9 or pT3T7lac.
In the case of the mutagenesis of the loop of the assumed stem-loop-structure, six separate fill-in reactions were performed. In each of the reactions one of six different oligonucleotides, each degenerate at one position of the loop, was used. It was annealed with three oligonucleotides providing the rest of the [DNA sequence] such that the site of degeneracy was situated in a single-stranded region.
To produce a SECIS with a mutated loop sequence, Heider starts by using oligonucleotide synthesis to create four short ssDNA (single-stranded DNA), where one of the four ssDNA contains the desired mutation. Each ssDNA has a base sequence that allows it to base pair with one or two of the other ssDNA, so that they can form a DNA with alternating single-stranded and double-stranded regions.
An example of such a DNA is shown below, where the loop sequence is marked with bold text. The wildtype SECIS-loop has the sequence AGGTCT, so this example DNA is a mutant where the first base of the loop is changed from A to G.
3'-TTCGAAAACTGTGCCGGGTAGCCAAp TGGTTAGCCAGCCAAAAACCTAGG-5'
The two oligonucleotides lying within the annealed product had been phosphorylated at their 5’-ends. The annealed product was treated simultaneously with T4 DNA polymerase and T4 DNA ligase to allow formation of complete double-stranded DNA.
The three single-stranded regions are filled in by T4 DNA polymerase and ligated by T4 ligase, so that a complete dsDNA (double-stranded DNA) is created. (T4 DNA polymerase originates from the T4 virus and is well suited for filling in gaps.)
The dsDNA after fill-in and ligation is shown below. The TGA codon (= UGA in mRNA) is underscored and the SECIS (selenocysteine insertion sequence) is marked with bold text.
3'-TTCGAAAACTGTGCCGGGTAGCCAACGCCCAGACGTGGTTAGCCAGCCAAAAACCTAGG-5'
In order for ligation to work, it is necessary that the 5'-ends being ligated are bound to a phosphate. DNA produced with oligonucleotide synthesis lacks phosphate at the 5'-ends, so the two short ssDNA which 5'-ends are to be ligated must first be phosphorylated. Phosphorylation is performed by mixing the DNA with ATP (adenosine triphosphate) and polynucleotide kinase, an enzyme capable of transferring phosphate from ATP to the 5'-end of DNA. In the example DNA with alternating single- and double-stranded regions (shown above), the phosphorylated 5'-ends are marked with "p".
In the case of mutagenesis of [the stem region of the stem-loop-structure], oligonucleotides were annealed pairwise and filled in using Klenow [DNA polymerase].
Mutations of the stem refers to the insertion or deletion of base pairs. Such mutations were created by using oligonucleotide synthesis to produce a long ssDNA, as well as a short ssDNA which can base pair to the 3'-end of the long ssDNA and function as a primer in the creation of a dsDNA. An example of such a DNA is presented below, where inserted bases are shown with bold text.
3'-TTCGAAAACTGTGCCCACGGGTAGCCAACGTCCAGACGTGGTTAGCCATGGGCCAAAAACCTAGG-5'
The missing nucleotides are filled in with DNA polymerase I (Klenow), which creates a complete dsDNA. The DNA after fill-in is presented below, where the UGA codon (TGA in DNA) is underscored and the SECIS (selenocysteine insertion sequence) is marked with bold text.
3'-TTCGAAAACTGTGCCCACGGGTAGCCAACGTCCAGACGTGGTTAGCCATGGGCCAAAAACCTAGG-5'
(Why the loop mutants and stem mutants are created with different methods, where the loop mutants are made from four short sequences and the stem mutants are made from one long and one short sequence, I do not know.)
When creating the loop mutants, T4 DNA polymerase was used to fill in the nucleotides and form a complete dsDNA, while in creating the stem mutants the nucleotides were filled in using DNA polymerase I (Klenow). Klenow cannot be used to fill in nucleotides in the loop mutants because Klenow has a "strand displacement activity", which causes the double-stranded regions of DNA to break apart from each other (see the figure below). T4 lacks the strand displacement activity and is therefore used to fill in the nucleotides in the loop mutants.
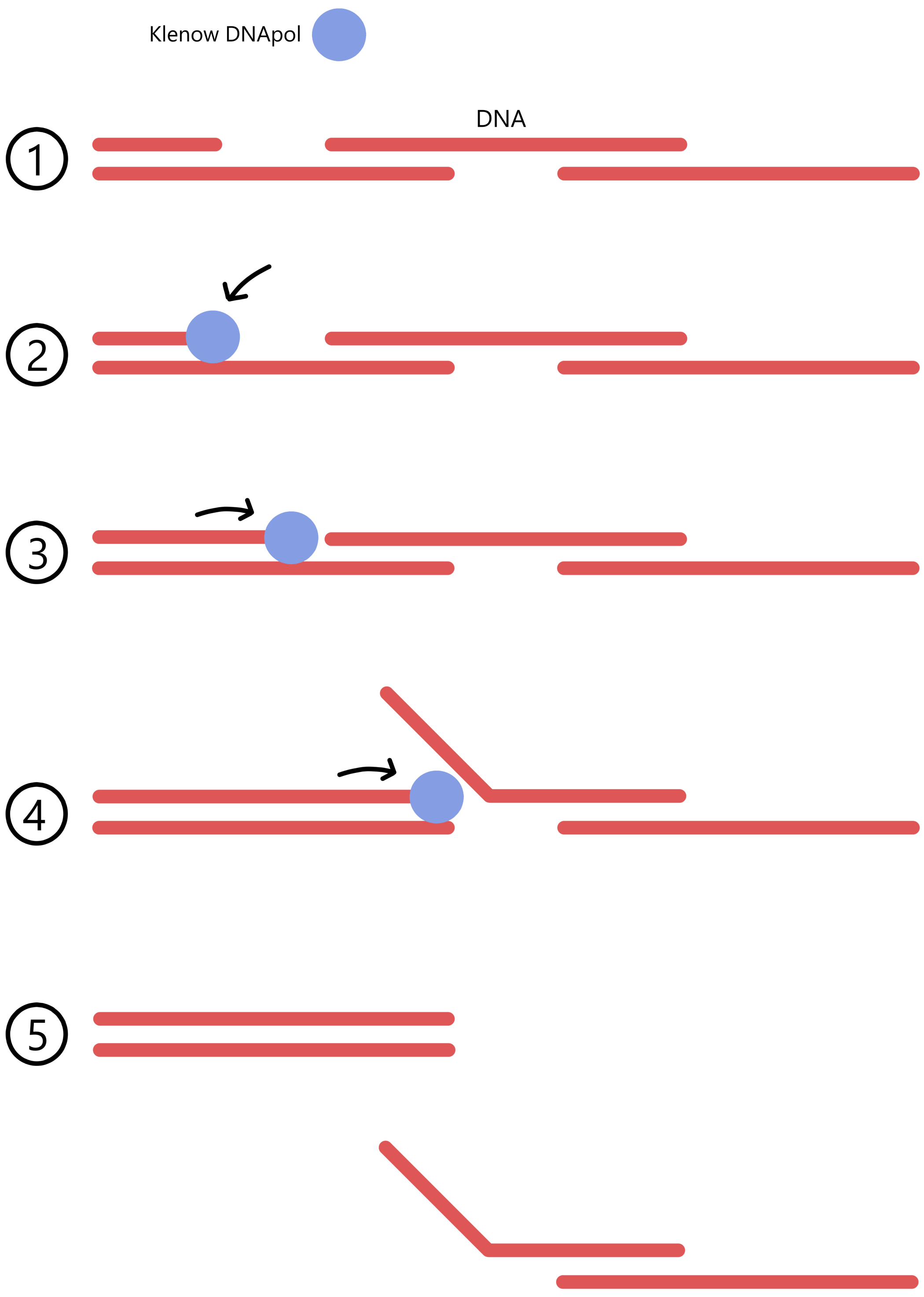
The stem mutants, on the other hand, were created using DNA polymerase I (Klenow) to fill in the nucleotides. (T4 DNA polymerase could also have been used here. I guess that Klenow is cheaper than T4, and that this is the reason why Klenow was used here.)
In both cases the DNAs were cut with HindIII and BamHI at restriction sites provided at their ends and inserted into plasmid pUC9 or pT3T7lac. After screening by sequencing, the wildtype and mutant DNA were inserted into plasmid pSKS106 and rechecked by sequencing.
All stem- and loop-mutants were made with a HindIII restriction site 5' of the SECIS, and a BamHI restriction site 3' of the SECIS.
/ \ / \
5'-AAGCTTTTGACACGGCCCATCGGTTGCGGGTCTGCACCAATCGGTCGGTTTTTGGATCC-3'
3'-TTCGAAAACTGTGCCGGGTAGCCAACGCCCAGACGTGGTTAGCCAGCCAAAAACCTAGG-5'
Plasmid pSKS106 contains a lacZ gene where a polylinker has been inserted between lacZ's fifth and sixth codon (according to Heider's reference: Shapira 1983). The SECIS mutants are inserted into the polylinker by using the restriction nucleases HindIII and BamHI. (I don't know why the mutants are inserted into plasmid pUC9/pT3T7lac before they are cut out and inserted into plasmid pSKS106.)
lacZ with inserted SECIS mutant is transcribed from the lacZ promoter and is translated from lacZ's start codon. The 5'-part of lacZ, which includes the SECIS mutant, is sequenced to verify that the correct DNA sequence has been correctly inserted into lacZ.
Synthesis of random mutants in Klug, Huttenhofer, Kromayer, Famulok (1997): In vitro and in vivo characterization of novel mRNA motifs that bind special elongation factor SelB
(This section requires an understanding of oligonucleotide synthesis.)
For his experiment, Klug produced SECIS-RNA with a random mutation rate of 30% ("30% degenerate insert", as he calls it). Creating randomly mutated RNA by means of oligonucleotide synthesis is simple. First one has to produce randomly mutated DNA, and then transcribe RNA from this DNA.
Here is a DNA containing the wildtype SECIS:
If this sequence is to be produced with oligonucleotide synthesis you'd start by using thymine-nucleotides, then guanines, then guanines again, then cytosine, then thymine, ... so that you get the sequence 3'-TGGCT... (In oligonucleotide synthesis the sequence is produced from the 3'-end towards the 5'-end.)
If, on the other hand, you're going to produce a SECIS with a random mutation rate of 30% you'd start by using a mixture of nucleotides, where there's 70% thymine-nucleotides and 30% other nucleotides (10% adenines, 10% cytosines, 10% guanines). Next you'd add a mix of 70% guanines and 30% other nucleotides. Then another mix of 70% guanines and 30% others. Then 70% cytosines and 30% others, then 70% thymines and 30% others, ...
This will lead to the production of a large number of different DNA sequences, where each base of the sequence has a 70% probability to match the SECIS wildtype and a 30% probability of being mutated. As mentioned in the section on Klug 1997, this means that the average DNA will have 12 mutated bases in the SECIS (39 SECIS-bases × 0.3 mutation rate ≈ 12 mutated bases), but since the process is random, many SECIS will have more or fewer than 12 mutations.
The DNAs produced by Klug didn't purely contain a SECIS, they also contained a T7 promoter, which together with the T7 RNA polymerase is used to transcribe the DNA to RNA. The transcribed SECIS-RNA is mixed with SelB proteins to investigate which SECIS-mutants can bind to SelB.
References
Kromayer, Wilting, Tormay, Bock
(1996): Domain Structure of the Prokaryotic Selenocysteine-specific Elongation Factor SelB. Paid article
Shapira, Chou, Richaud, Casadaban
(1983): New versatile plasmid vectors for expression of hybrid proteins coded by a cloned gene fused to lacZ gene sequences encoding an enzymatically active carboxy-terminal portion of beta-galactosidase. Paid article
Smeal, Schmitt, Pereira, Prasad, Fisk
(2016): Simulation of the M13 life cycle I: Assembly of a genetically-structured deterministic chemical kinetic simulation. Free article