Fotometri i biologi
© Anders Skovly 2025
(Denne teksten fokuserer hovedsakelig på det teoretiske aspektet til fotometri, ikke på metodens praktiske anvendelser.)
I min artikkel om selenocystein-innsettingselementet ble det nevnt hvordan en metode kalt fotometri ble brukt både for å måle konsentrasjonen av molekylet o-nitrophenol i en prøve, og for å måle konsentrasjonen av celler i en prøve. Å måle cellekonsentrasjon via fotometri er en vanlig metode når en arbeider med bakterier og andre encellede organismer, så det var derfor et naturlig emne å skrive om her. Mer nøyaktig så vil denne artikkelen dekke følgende:
Del 1: Det grunnleggende
Hvordan et fotometer fungerer
Et fotometer er en maskin som måler gjennomsiktigheten til en prøve (et test-objekt). Innen celle- og molekylærbiologi vil prøven være en væske som inneholder celler eller molekyler. Når en måling skal utføres med et fotometer, så blir et volum av en væske først overført til en liten gjennomsiktig beholder kalt en kyvette. Inni fotometeret finnes en kyvette-holder hvor kyvetten settes inn. Se dette bildet for et eksempel (dette fotometeret har to kyvette-holdere).
{kind=link}
På en side av kyvette-holderen er det en lyskilde, og på den andre siden er det en lyssensor. Mellom lyskilden og kyvetten finnes en komponent kalt en bølgelengde-velger. Når en bruker et fotometer er det viktig å velge korrekt bølgelengde for prøven man måler. Valg av bølgelengde vil bli beskrevet senere i artikkelen. For en illustrasjon av oppsettes av fotometerets komponenter, se Graphic 2 i denne teksten.
Lyset som kommer fra lyskilden, går gjennom bølgelengde-velgeren og treffer prøven kalles “innfallende lys” (incident light). Intensiteten (lysstyrken) til innfallende lys symboliseres normalt med I0. Lyset som passerer gjennom prøven og treffer lyssensoren kalles “transmittert lys”. Intensiteten av transmittert lys vil symboliseres i denne teksten med Is (hvor “s” står for “sample”, altså det engelske ordet for “prøve”).
Fotometeret måler andelen av transmittert lys til innfallende lys (Is / I0) og presenterer denne andelen i form av prøvens “transmittans”, symbolisert i denne teksten med Ts. Altså,
Ts = Is / I0
(Intensiteten av innfallende lys (I0) kan bestemmes ved å ikke plassere noen kyvette/prøve i kyvette-holderen og dermed la lyskilden lyse direkte på lyssensoren.)
Den høyeste mulige verdien av transmittans er 1, eller 100%. Denne verdien indikerer at alt innfallende lys kan gå rett gjennom prøven (noe som egentlig kun vil skje dersom ingen prøve er plassert i kyvette-holderen). Den laveste mulige verdien av transmittans er 0, som indikerer at prøven er fullstendig ugjennomsiktig slik at ingen lysstråler kan passere gjennom.
Eksempel-måling og beregning
La oss si at vi har en kultur av bakterien Aeromonas salmonicidia som vokser i et flytende medium, og at vi ønsker å vite konsentrasjonen av A. salmonicidia-celler i mediet. (“Cellekonsentrasjon” betyr antallet celler per volum-enhet, for eksempel antallet celler per milliliter medium.) Her er en metode for å bestemme konsentrasjonen ved bruk av et fotometer:
Først, mål intensiteten av innfallende lys (I0) ved å trykke på fotometerets “calibrate” eller “zero”-knapp mens kyvetteholderen er tom.
Deretter, overfør et lite volum av medium til en kyvette (rent medium UTEN bakterier). Dette kalles en blank. Putt kyvetten inn i kyvetteholderen og trykk på fotometerets “measure”-knapp for å måle intensiteten av lyset som passerer gjennom blank-prøven (Ib). Fotometeret vil beregne verdien av Ib/I0 og vise resultatet som blank-prøvens transmittans (Tb).
Til sist, hell ut det rene mediumet og fyll kyvetten opp igjen med et lite volum av bakteriekulturen. Putt kyvetten inn i kyvetteholderen og trykk på fotometerets “measure”-knapp for å måle intensiteten av lyset som passerer gjennom prøven (Is). Fotometeret beregner verdien av Is/I0 og viser resultatet som prøvens transmittans (Ts).
Nå har vi utført alle nødvendige målinger og har verdiene for Ts og Tb. Videre må vi forstå begrepet optisk tetthet (optical density, OD). Optisk tetthet er logaritmen av resiprokalen av transmittansen, altså log10(1/T). Derfor er prøvens optiske tetthet log10(1/Ts), og blank-prøvens optiske tetthet er log10(1/Tb). (Heretter vil “log10” kun skrives som “log”.)
Dersom transmittans er 1 vil optisk tetthet være 0 (log(1/1) = 0). Lavere transmittans korresponderer til høyere optiske tettheter. Se tabellen nedenfor for relasjoner mellom gitte transmittans-verdier og optisk tetthet-verdier.
| Transmittans (T) | Optisk tetthet (OD, log(1/T)) |
|---|---|
| 1.00 | 0.00 |
| 0.90 | ≈0.05 |
| 0.75 | ≈0.12 |
| 0.50 | ≈0.30 |
| 0.25 | ≈0.60 |
| 0.10 | 1.00 |
| 0.01 | 2.00 |
En prøves optiske tetthet (OD) kan anses å ha to additive komponenter: OD som kommer fra kyvetten og mediet, og OD som kommer fra bakteriecellene:
log(1/Ts) = ODs = ODkyvette,medium + ODceller
En blankprøves optiske tetthet kan anses å ha kun en komponent: OD som kommer fra kyvetten og mediet:
log(1/Tb) = ODb = ODkyvette,medium
Derfor, hvis vi subtraherer log(1/Tb) fra log(1/Ts) så får vi verdien av ODceller:
log(1/Ts) – log(1/Tb)
= (ODkyvette,medium + ODceller) – (ODkyvette,medium)
= ODceller
Konsentrasjonen av A. salmonicidia i prøven er proporsjonal med ODceller og kan beregnes fra en ligning kalt Beer's lov:
c × p × ε = log(1/Ts) – log(1/Tb) = ODceller
hvor c er cellekonsentrasjonen, p er "path length", og ε er ekstinksjonskoeffisienten. (Ordet “ekstinksjon” refererer til reduksjonen i lysintensitet som skjer når lyset går gjennom prøven.)
“Path length” refererer til distansen som lyset går gjennom prøven. Den vanligste typen kyvette har et “kvadratisk hull” som måler 1 cm ganger 1 cm (her er et bilde som viser dette). Derfor, når en slik kyvette er fylt med bakteriekultur, så vil kulturen fylle et volum som måler 1 cm bredt. Dette betyr at når kyvettes settes inn i et fotometer så vil lyset som entrer kyvetten gå gjennom 1 cm bakteriekultur før lyset kommer ut av kyvetten. Altså er lysets “path length” 1 cm i en slik kyvette.
Ekstinksjonskoeffisienten ε avhenger både av cellene hvis konsentrasjon en måler, og av det fotometeret som benyttes for å måle transmittansen (senere seksjoner vil gi mer detaljer på dette). Dersom man allerede har funnet ekstinksjonskoeffisienten, så er fotometri er rask og enkel metode for å beregne cellekonsentrasjoner. På en annen side, dersom en ikke kjenner ekstinksjonskoeffisienten, så vil dens bestemmelse kreve at vi lager en standardkurve.
(Jeg burde egentlig inkludere en eksempel-beregning her, hvor c beregnes ut ifra realistiske verdier for ε, log(1/Ts) og log(1/Tb). Men ettersom jeg for tiden ikke har tilgang på et fotometer og ikke kan skaffe slike verdier akkurat nå, så må det vente til senere.)
(Noen fotometre viser transmittans ikke som en brøkverdi mellom 0 og 1, men som en prosent-verdi mellom 0% og 100%. Dersom fotometeret gir verdier som % transmittans, så må prosent-verdiene deles med 100 før de settes inn i Beer's lov.)
Forenklet eksempel-måling og beregning
Ligningen for Beer's lov som ble presentert i forrige seksjon er ikke den ligningen som normalt benyttes. Normalt brukes istedet en enklere (men matematisk likeverdig) ligning:
c × p × ε = log(1/T) = ODceller
hvor “T” representerer brøken Is/Ib. Jeg viste den andre, mer “komplekse” ligningen først fordi den er mer intuitiv. Dette er hvordan den forenklede ligningen deriveres fra den første:
Begynn med
log(1/Ts) – log(1/Tb)
og substituer transmittanser med lysintensiteter for å få
log(I0/Is) – log(I0/Ib)
Ifølge logaritme-kvotient-regelen er dette uttrykket det samme som
log( I0/Is / I0/Ib )
De to I0 kansellerer hverandre, sånn at uttrykket kan skrives som
log( 1/Is / 1/Ib )
Ved å multiplisere begge sider av brøken med (Is × Ib) får vi
log(Ib/Is)
Ettersom T = Is/Ib, så har vi også at 1/T = Ib/Is, sånn at uttrykket ovenfor blir til
log(1/T)
Som er det uttrykket som brukes i den forenklede Beer's lov. Sammen med den forenklede ligningen har vi en forenklet metode:
Først, overfør et lite volum medium til en kyvette (rent medium UTEN bakterier). Dette er blankprøven. Putt kyvetten i kyvetteholderen og trykk på fotometerets “calibrate” eller “zero”-knapp for å måle intensiteten av lyset som går gjennom blankprøven (Ib).
Deretter, hell ut det rene mediet og fyll kyvetten med et lite volum av bakteriekultur. Putt kyvetten i kyvetteholderen, og trykk på fotometerets “measure”-knapp for å måle intensiteten av lyset som går gjennom prøven (Is). Fotometeret beregner verdien av Is/Ib og viser resultatet i form av transmittansen “T”, som er den eneste transmittansen som trengs i den forenklede Beer's lov.
Hvordan lage en standard-kurve
En standard-kurve brukes til å bestemme verdien av ekstinksjonskoeffisienten ε i Beer's lov. Si at vi har tre bakteriekultur-prøver: prøve 1, prøve 2, og prøve 3. De tre prøvene har tre ulike cellekonsentrasjoner c1, c2, and c3. Vi kjenner verdiene av de tre konsentrasjonene, da de har blitt bestemt med en metode alternativ til fotometri (for eksempel plate-telling-metoden). Vi har også en blankprøve, rent medium uten bakterier.
Et fotometer brukes for å beregne log(1/T) for hver av de tre prøvene, slik at vi får de tre verdiene log(1/T1), log(1/T2), og log(1/T3). Ettersom
c × p × ε = log(1/T)
så har vi også at
c = 1/(p × ε) × log(1/T)
som betyr at c har en lineær relasjon til log(1/T).
La hvert par av konsentrasjon og log(1/T), slik som paret av c1 og log(1/T1), definere et punkt i et x-y-plan, med log(1/T) på x-aksen og konsentrasjonen på y-aksen. Når de tre punktene plottes i x-y-planet vil de ligge langs en rett linje. Stigningstallet til denne linjen har verdien 1/(p × ε).
For å bestemme stigningstallet nøyaktig bruker vi lineær regresjon for å finne ligningen for linjen som går gjennom de tre punktene. I denne regresjonen vil log(1/T) være input-variabelen, og konsentrasjonen vil være den predikerte variabelen. Som med alle rette linjer så vil vår lineære regresjonslinje ha den generelle formelen y = ax + b. I vårt tilfelle, hvor x er log(1/T) og y er konsentrasjonen, så får vi en regresjonslinje med formelen c = a × log(1/T) + b. Verdiene for “a” and “b” bestemmes av regresjonsanalysen. Verdien for “a” er et estimat av 1/(p × ε).
I en prøve uten bakterie (altså en prøve hvor cellekonsentrasjonen er 0) skal log(1/T) ha en verdi på 0 (fordi når c = 0 så er Is = Ib, slik at transmittansen T = 1, og log(1/T) = log(1/1) = 0). Dersom vi setter c = 0 og log(1/T) = 0 inn i regresjonsformelen så får vi 0 = a × 0 + b. Derfor skal b i teorien være 0.
Som følge av unøyaktigheter i bestemmelsen av cellekonsentrasjoner, og som følge av unøyaktigheter i fotometerets bestemmelse av log(1/T), så vil regresjonsligningen mest sannsynlig ha en "non-zero" verdi av b. Men, dersom cellekonsentrasjoner og log(1/T) har blitt bestemt relativt nøyaktig, så skal verdien av b være nær 0.
(Igjen, det ville vært passende å utføre en eksempel-regresjon med realistiske verdier av c og log(1/T) for å vise hvordan ε bestemmes, men siden jeg ikke har tilgang på et fotometer nå så kan det ikke gjøres.)
Regresjonslinjen og dens ligning blir vanligvis referert til som en “standard-kurve”. Dette fordi kurven (egentlig ikke en kurve men en linje i dette tilfellet) lages ved å bruke prøver med kjente konsentrasjoner. Slike prøver kalles “konsentrasjons-standarder”.
Hvordan bruke en standard-kurve
La oss si at vi har en fjerde bakteriekultur hvor vi ikke kjenner cellekonsentrasjonen. For å bestemme konsentrasjonen kan vi nå bruke fotometeret til å bestemme log(1/T) for en kulturprøve, putte verdien inn i standard-kurve-ligningen c = a × log(1/T) + b, og så beregne verdien av c.
Det må understrekes at den beregnede konsentrasjons-verdien kun er et estimat, hvor nøyaktigheten av estimatet avhenger av hvor nøyaktig fotometeret målet transmittans, og av hvor nøyaktig vi kjenner cellekonsentrasjonene som benyttes til å lage standard-kurven. (“Nøyaktig” betyr hvor nær en målt verdi er til den ekte verdien.)
Del 2: Absorbans, optisk tetthet, og bølgelengder
Bølgelengde, farge, og absorbans
Så langt har vi sett hvordan et fotometer fungerer og hvordan det kan brukes sammen med Beer's lov for å bestemme cellekonsentrasjonen i en prøve. Senere i artikkelen vil bestemmelse av cellekonsentrasjon bli utdypet, men la oss nå bytte til et nytt tema: hvordan beregne konsentrasjonen av et lys-absorberende molekyl i en løsning. Vi starter dette tema med en seksjon om bølgelengde, farge, og absorbans.
Hvitt lys, enten fra solen eller fra en typisk lyspære (LED eller glødetråd), inneholder lys av alle farger i regnbue-spekteret: rød, oransje, gul, grønn, blå, og fiolett. Når lys av disse fargene sees sammen ser lyset hvitt ut.
Alt lys består av elektromagnetiske bølger. Denne teksten er ikke det riktige stedet for å forklare hva det betyr at lys er elektromagnetiske bølger, men det er en detalj som gjør det viktig å nevne dette. Når en refererer til en bestemt farge i regnbuespekteret kan fargen refereres til med dens “bølgelengde”. Bølgelengde i denne konteksten er et nummer som måles i nanometer, eller nm (en nanometer er 10-9 meter, altså en milliard-del av en meter). For å forstå hvordan bølgelengder korresponderer til ulike farger i regnbuespekteret, se denne figuren.
{kind=link}
Som figuren viser så befinner regnbuespekteret seg i regionen fra omtrent 400 nm til 700 nm. 400 nm korresponderer til lys av fiolett farge, mens 700 nm korresponderer til lys av rød farge. Det finnes lys under 400 nm og over 700 nm, men slikt lys kan ikke oppfattes av det menneskelige øyet og har derfor ingen farge. Under 400 nm bølgelengde har vi ultrafiolett lys (UV-lys), mens over 700 nm bølgelengde har vi infrarødt lys.
Jeg nevnte i begynnelsen av denne artikkelen at fotometri kan brukes til å måle konsentrasjonen av molekylet o-nitrophenol i en prøve. En løsning av o-nitrophenol har en gul farge, som en kan se i dette bildet.
{kind=link}
Hvorfor er o-nitrophenol gult? For å begynne å besvare dette spørsmålet, la oss tenke på hva som skjer når en løsning av o-nitrophenol belyses av hvitt lys bestående av alle regnbuespekterets farger. Hver lysstråle kan enten gå rett gjennom løsningen, eller den kan treffe et av molekylene av o-nitrophenol som er jevnt spredt i løsningen. (Molekylene kan visualiseres som små partikler som flyter rundt). Dersom en lysstråle treffer en o-nitrophenol er det en sjanse for at strålen blir absorbert av o-nitrophenolen. For enkelhets skyld kan vi tenke at absorbsjon betyr at lysstrålen forsvinner, den finnes ikke lenger.
Ikke alle av lysets farger absorberes like effektivt. Ta en titt på Figur 2 i denne teksten. Den lyseblå kurven viser absorbans-spekteret til o-nitrophenol i en løsning med høy pH (omtrent pH 11, tror jeg). Spekteret viser at o-nitrophenol absorberer lys med 420 nm bølgelengde (fiolett lys) mer effektivt enn andre bølgelengder. Absorbsjon blir mindre effektiv desto lenger unna en bølgelengde er fra 420 nm. Det sies at o-nitrophenol har en “absorbans-topp” ved 420 nm.
Altså, når en løsning av o-nitrophenol belyses av hvitt lys, så vil o-nitrophenol hovedsakelig absorbere fiolett lys, og det transmitterte lyset (altså lyset som går gjennom løsningen) ser gult ut. Gul er den komplementære fargen til fiolett. Det er en generell regel at dersom et molekyl absorberer en bestemt farge av lyset, så vil det transmitterte lyset ha den komplementære fargen av den absorberte fargen. (For mer informasjon, see britannica.com/science/color, seksjonen kalt “The laws of colour mixture”.)
Bølgelengde-velgeren
I Del 1 av denne artikkelen forklarte jeg hvordan Beer's lov kan brukes til å finne konsentrasjonen av bakterieceller i en prøve. Men det var en viktig detalj som ikke ble nevnt i Del 1, nemlig valget av fotometerets bølgelengde. La oss nå se på hva en bølgelengde-velger er for noe.
Et fotometer inneholder en kyvetteholder med en lyskilde på den ene siden og en lyssensor på den andre siden. Lyskilden sender ut hvitt lys bestående av alle fargene i regnbuespekteret. Mellom lyskilden og kyvetteholderen finnes en komponent kalt en bølgelengde-velger (wavelength selector), som lar brukeren velge en bestemt bølgelengde fra lyskilden som transmitteres til prøven i kyvetteholderen. Alle de andre bølgelengdene i det hvite lyset blokkeres av bølgelengde-velgeren.
I noen fotometre er bølgelengde-velgeren basert på lysfiltre. Hvilke bølgelengder som kan velges er da avhengig av hvilke filtre som er tilgjengelige. For eksempel, dersom du kun har et 420 nm filter og er 600 nm filter, så kan du kun velge en av disse to bølgelengdene. 420 nm-filteret absorberer alt lys utenfor 420 nm-regionen, slik at kun lys i 420 nm-regionen transmitteres til prøven. Og 600 nm-filteret absorberer alt lys utenfor 600 nm-regionen, slik at kun lys i 600 nm-regionen transmitteres til prøven.
I andre fotometre er bølgelengde-velgeren basert på et diffraksjons-gitter (diffraction grating) sammen med en “utgangs-sprekk” (exit slit). Se hvordan et diffraksjonsgitter fungerer her og her. Fordelen med et diffraksjonsgitter over et filter er at fotometeret kan rotere diffraksjonsgitteret for å justere hvilken bølgelengde som passerer gjennom utgangssprekken. Gitteret kan dermed velge en hvilken som helst bølgelengde innen et bestemt område, med små intervaller mellom hver valgbar bølgelengde. For eksempel, fotometeret som jeg brukte på universitetet, en Hitachi U-5100, kan velge en hvilken som helst bølgelengde i området fra 190 nm til 1100 nm, i intervaller på 0.1 nm.
{kind=link}
(Det finnes også bølgelengde-velgere som er basert på prismer, men slik jeg forstår det så er ikke prism-baserte bølgelengde-velgere vanlig lenger.)
Hverken et filter eller et diffraksjonsgitter vil blokkere alt lys utenfor den valgte bølgelengden. Istedet vil filteret eller gitteret transmittere et “bånd” av bølgelengder (en gruppe av lignende bølgelengder). Bølgelengde-båndet vil være sentrert på den valgte bølgelengden. Denne figuren viser et eksempel på et bølgelengde-bånd sentrert på 710 nm.
{kind=link}
(Merk at et bølgelengde-bånd ikke alltid vil være “bjelle-formet” slik som kurven i figuren over. Formen på bølgelengde-båndet avhenger av emisjonsspekteret til fotometerets lyskilde.)
Bredden av bølgelengde-båndet kalles båndbredden, og måles vanligvis som “full width at half maximum” (FWHM). Dette er bredden av bånd-kurven målt halvveis mellom baselinjen og kurvens toppunkt. Båndet i figuren ovenfor har en båndbredde på 10 nm (målt fra 705 nm til 715 nm).
Måling av konsentrasjon av o-nitrophenol
Når du bruker et fotometer for å måle transmittans vil du vanligvis ikke bruke transmittansen direkte, ihvertfall ikke hvis du jobber innen biologi. Istedet vil du vanligvis bruke log(1/T). Istedet for å ta logaritmen av transmittansen manuelt kan du derfor sette fotometeret til å vise resultater i form av log(1/T) istedet for T.
Når en måler log(1/T) i en løsning av lys-absorberende molekyler, så blir log(1/T) referert til som “absorbansen” til løsningen. Dette er en viktig detalj, fordi dersom du vil bytte fotometeret fra å vise transmittanser til å vise log(1/T), så vil det ikke være et valg som heter “log(1/T)”. Istedet vil det være et valg som heter “Absorbance” eller “Abs”. Hvis du velger dette vil fotometeret vise verdier i form av log(1/T).
(Dette er ihvertfall sant for Hitachi U-5100, at du må velge “Abs” for å få log(1/T). Det kan være forskjellig på andre fotometre, men jeg tror det er vanlig at fotometre refererer til log(1/T) som “Abs”.)
La oss si at vi har en løsning (pH 11) med en ukjent konsentrasjon av o-nitrophenol, og at vi ønsker å bestemme konsentrasjonen. Først setter vi fotometeret til å vise absorbans. Deretter velger vi bølgelengden. Når en måler absorbansen av lys-absorberende molekyler, så velger man normalt den bølgelengden som korresponderer til absorbans-kurvens toppunkt. Som beskrevet i en tidligere seksjon, i en løsning med pH 11 har o-nitrophenol en absorbans-topp ved 420 nm (se Figur 2 her). Derfor stiller vi fotometeret til å bruke en bølgelengde på 420 nm.
Som når vi målte konsentrasjonen av bakterieceller, så setter vi en blankprøve i fotometeret (blankprøven i dette tilfellet er en kyvette fult med løsemiddel, UTEN o-nitrophenol). Trykk på “zero” eller “calibrate”-knappen på fotometeret. Hell så ut løsemiddelet og fyll kyvetten med løsningen av o-nitrophenol, sett kyvetten inn i fotometeret igjen og trykk på “measure”-knappen. Verdien av absorbans (log(1/T)) som vises brukes så i Beer's lov for å beregne konsentrasjonen av o-nitrophenol i løsningen. (Dette krever at du allerede har en standard-kurve for o-nitrophenol, som gir deg verdien av 1/(p × ε) som trengs i Beer's lov.)
Turbiditet og lysspredning
Nå skifter vi tema tilbake til celler. Når celler vokser i en flytende kultur så blir kulturen turbid, eller “cloudy”. Graden av turbiditet avhenger av konsentrasjonen av celler, hvor høyere cellekonsentrasjoner gir høyere turbiditet. Se for eksempel dette bildet, hvor tuben til venstre har høyest turbiditet og derfor høyest cellekonsentrasjon.
Når en lysstråle treffer en celle i en flytende kultur vil lysstrålen avbøyes i vinkel, slik at den vil bevege seg i en ny retning. Forskjellige lysstråler avbøyes i forskjellige retninger, slik at lyset blir spredt (scattered). Fenomenet med lysspredning er årsaken til turbiditet. Her er en figur som viser lysspredning.
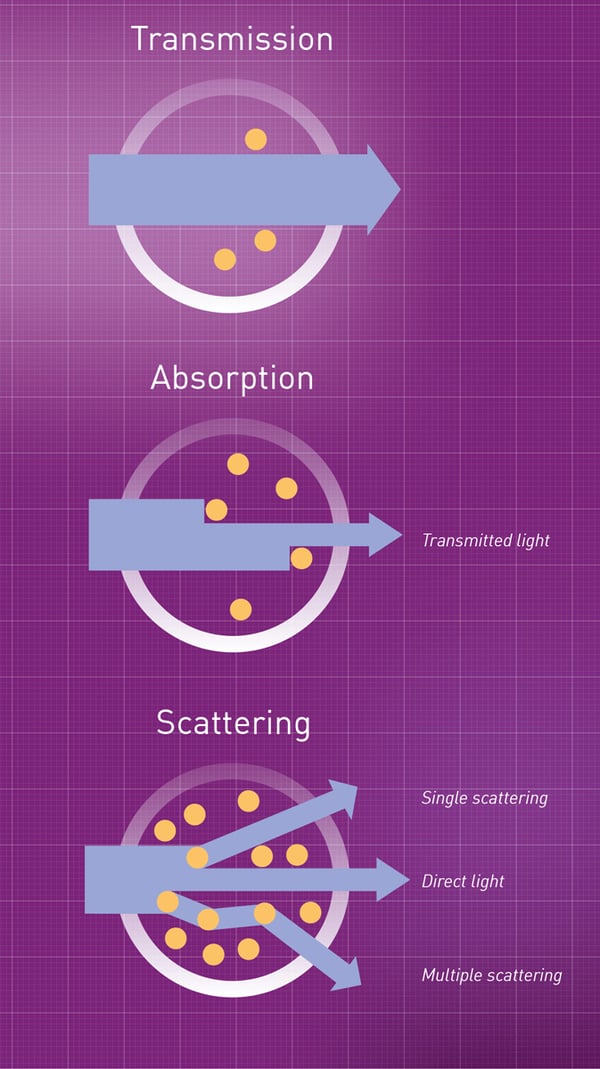{kind=link}
Når vi bruker et fotometer for å måle konsentrasjonen av et lys-absorberende molekyl i en prøve, så vil reduksjonen i transmittans (relativt til blankprøven) være forårsaket av at noen av lysstrålene absorberes av molekylene. Disse lysstrålene “forsvinner” og vil derfor ikke treffe lyssensoren. I motsetning, når vi bruker fotometeret til å måle cellekonsentrasjonen i en prøve, så vil reduksjonen i transmittans være forårsaket av at noen lysstråler avbøyes og spres av cellene, slik at de endrer retning bort fra lyssensoren. Disse lysstrålene transmitteres fremdeles gjennom den flytende kulturen, men de treffer ikke sensoren og blir derfor ikke registrert.
Måling av cellekonsentrasjon (igjen)
Bruk av et fotometer for å måle log(1/T) og beregne cellekonsentrasjon i en kultur har allerede blitt forklart tidligere i artikkelen, så nå vil jeg bare gi noen ekstra kommentarer. Først, bølgelengden som brukes til å måle log(1/T) i en cellekultur er vanligvis 600 nm.
Og en annen ting, dersom du vil at fotometeret skal vise verdier i form av log/1/T), så må du sette fotometeret til å vise verdiene i form av “Abs”. Dette til tross for at du ikke måler lysabsorpsjon. Det faktum at du måler lysspredning og ikke lysabsorpsjon er ikke av betydning, fordi “Abs” er kun det navnet fotometeret bruker for log(1/T), som er det du vil måle. (Fotometre er lagd for å måle transmittans av lys-absorberende molekyler i løsning, som er grunnen til at fotometre refererer til log(1/T) som “Abs”. Å bruke fotometeret til å måle lysspredning er en “alternativ bruk”.)
Når vi måler cellekonsentrasjon i en kultur, så blir verdien av log(1/T) istedet referert til som kulturens “optiske tetthet” (optical density, OD). (Det kan være tilfeller hvor vi måler transmittans av en prøve som inneholder både celler og lys-absorberende molekyler, slik at både absorpsjon og lysspredning bidrar til reduksjon av transmittans. I slike tilfeller vil log(1/T) refereres til som optisk tetthet.)
(Bare et lite historie-notat: det ser ut til at fotometrisk måling av cellekonsentrasjoner først ble brukt etter 1935. Monod 1949 skriver: A few general remarks may be made about the techniques employed for the estimation of bacterial density and cell concentrations [...] The most widely used methods, by far, are based on determinations of transmitted or scattered light. (Actually, the introduction around 1935 of instruments [photometers] fitted with photoelectric cells [light sensors] has contributed to a very large degree to the development of quantitative studies of bacterial growth.) [...] The instruments best fitted for the purpose appear to be those which give readings in terms of optical density (log I0/I).)
Del 3: Betraktninger ved bruk av fotometri
Fotometeret kan ikke skille mellom levende og døde celler
Når en måler konsentrasjonen av celler i en prøve, så er en vanligvis kun interessert i antallet levende celler: de som kan lage nye proteiner, vokse og dele seg til nye celler. En er gjerne ikke interessert i antallet døde celler i en prøve. Når en måler cellekonsentrasjon med et fotometer må en huske at fotometeret måler konsentrasjonen av levende og døde celler samlet, ettersom fotometeret ikke kan skille de levende cellene fra de døde (både levende og døde celler sprer lyset).
(Mengden døde celler som en andel av alle celler i en kultur er minimal i den eksponensielle vekstfasen. I denne vekstfasen deler cellene seg raskt, og mediet har fremdeles rikt med næringsstoffer. Ettersom kulturen skifter fra den eksponensielle vekstfasen til den stasjonære fasen, så begynner næringsstoffene å bli oppbrukt og mengden døde celler vil begynne å akkumulere.)
Beer's lov gjelder kun for lave konsentrasjoner
Figur 1 i Myers 2013 viser relasjonen mellom målt absorbans/OD og “sann absorbans/OD” for lys-absorberende molekyler (Fig 1A) og for E. coli-celler (Fig 1B). “Målt absorbans/OD” er verdiene gitt av fotometeret, mens “sann absorbans/OD” i praksis mener konsentrasjonen av celler/molekyler.
Figuren viser at når konsentrasjonen er lav, så er absorbans/OD lineær med konsentrasjonen, mens når konsentrasjonen er høy, så er det ikke lenger en lineær relasjon mellom absorbans/OD og konsentrasjon. Dette betyr at Beer's lov ikke gjelder ved høye konsentrasjoner, den gjelder kun for lave konsentrasjoner.
I tilfellet med lys-absorberende molekyler, så gjelder den lineære relasjonen for konsentrasjoner som tilsvarer en målt absorbans på 2 eller lavere. To grunner til at den lineære relasjonen ikke gjelder ved høyere absorbanser er elektrisk støy i fotometerets elektriske kretser, og strø-lys (stray light) som når lyssensoren. (Jeg vil ikke prøve å utdype disse to konseptene.)
I tilfellet med E. coli-celler så gjelder den lineære relasjonen for cellekonsentrasjoner som korresponderer til en målt OD på 0.5 eller lavere. Hovedårsaken til at den lineære relasjonen ikke gjelder ved høyere konsentrasjoner er såkalt “multiple scattering”. Dette refererer til en hendelse hvor en lysstråle treffer en celle, avbøyes bort fra lyssensoren, treffer en andre celle, og avbøyes på nytt. I noen tilfeller vil lys som avbøyes på nytt bli avbøyd tilbake mot lyssensoren. Frekvensen for multiple scattering øker med økende cellekonsentrasjoner. Relasjonen mellom cellekonsentrasjon og lys som avbøyes bort fra sensoren blir derfor ikke opprettholdt når konsentrasjonen blir for høy.
Det finnes måter for å håndtere problemet med ikke-lineæritet. Den vanligste løsningen er å fortynne prøven til en lavere konsentrasjon, måle absorbans/OD for denne lavere konsentrasjonen, og så multiplisere absorbans/OD med fortynningsfaktoren for å få den “sanne absorbans/OD”. For eksempel, dersom du har en prøve med celler hvor OD måler 1.0, så kan du fortynne prøven 1/3, måle OD for den fortynnede prøven (målt OD skal nå være under 0.5), og så multiplisere målt OD med 3 for å få den “sanne OD”, som så kan brukes til å beregne cellekonsentrasjonen med Beer's lov.
En annen måte for å omgå problemet er å måle absorbans/OD i en kyvette med kortere path length, da dette også reduserer målt absorbans/OD. Å redusere path length fra den normale 1 cm ned til 0.5 cm vil redusere målt absorbans/OD til omtrent halv verdi. Når en så bruker Beer's lov til å beregne konsentrasjon må den reduserte path length settes inn i ligningen i stedet for den standard 1 cm path length.
Selv om det er mulig å lage en standard-kurve fra kun tre konsentrasjons-standarder, så kan det være bedre å benytte et større antall konsentrasjons-standarder, da dette vil gjøre det mulig å se hvor høy konsentrasjonen kan være før den lineære relasjonen mellom konsentrasjon og log(1/T) ikke holder. Datapunkter som, når de plottes, viser tydelig ikke-lineæritet kan forkastes. Kun de datapunktene som viser lineæritet brukes i den lineære regresjonen for å lage standard-kurve-ligningen.
Målt absorbans/OD varierer mellom ulike fotometre
Figur 4 i Myers 2013 viser at en gitt konsentrasjon av krystallfiolett (et farget molekyl) har ulike absorbans-verdier når det måles på ulike fotometre. Og en gitt konsentrasjon av E. coli-celler har ulike OD-verdier når det måles på ulike fotometre. Variasjonen i målt OD for E. coli-prøven er større enn variasjonen i målt absorbans for krystallfiolett-prøven.
Ettersom forskjellige fotometre gir forskjellige verdier for den samme prøven, så er det nødvendig at målinger som gjøres for å lage en standard-kurve, samt senere målinger for å beregne konsentrasjoner ut ifra standard-kurven, alle må utføres på samme fotometer. Målinger fra ulike fotometre bør generelt ikke sammenlignes med hverandre.
Angående årsaken for ulikheter i absorbans/OD, la oss starte med ulikheter i absorbans. Fotometre kan bruke forskjellige lyskilder, hvor hver lyskilde har sitt eget emisjonsspektrum. Vanlige lyskilder er deuterium-lamper, tungsten-halogen-lamper, og xenon flash-lamper. Emisjonsspektra fra disse lampene kan ses i Figurer 9, 10, og 11 i denne teksten.
Videre, når en setter fotometeret til å måle ved en bestemt bølgelengde, si 420 nm, så treffes ikke prøven kun av lys med 420 nm bølgelengde. Istedet treffes prøven av et bølgelengde-bånd sentrert på 420 nm. Ulike lyskilder, i kombinasjon med ulike konstruksjoner av bølgelengde-velgere, gir bånd-kurver med forskjellige bredder og fasonger. (Dette er ikke helt enkelt å forstå, så noen figurer for å illustrere ulike bånd-kurver hadde vært passende, men jeg har ingen figurer som kan illustrere dette skikkelig.)
Når komposisjonen av innfallende lys varierer mellom fotometre, så vil fraksjonen av lys som absorberes og fraksjonen som transmitteres også variere mellom fotometre. Dette fører til ulikheter i målt absorbans.
Variasjon i målt OD for en celleprøve er også forårsaket delvis av ulike lyskilder og bølgelengde-velgere, men en annen faktor er størrelsen på lyssensoren, og sensorens posisjon relativt til prøven. Et eksempel på dette vises i Figur 3 på denne nettsiden. Når en lysstråle treffer en celle, la oss tenke at lyset enten kan avbøyes i en “liten vinkel”, en “middels vinkel”, eller en “stor vinkel”, relativt til den rette linjen mot sensoren. (I virkeligheten kan lyset avbøyes i mange ulike vinkler, men her forenkler vi det til kun tre vinkel-kategorier.)
Si at vi har to forskjellige fotometre, et med en stor lyssensor posisjonert nært prøven, og et med en liten sensor posisjonert lengre unna prøven. Begge sensorene registrerer alt lys som går rett igjennom prøven. Og begge sensorene vil registrere alt lys som kun avbøyes i en liten vinkel. Lys som avbøyes i en middels vinkel vil treffe den store, nære sensoren, men vil ikke treffe den mindre, fjærne sensoren. Og lys som avbøyes i en stor vinkel treffer hverken av sensorene. Forskjeller i hvor mye spredt lys som kan registreres av sensorene fører til variasjon i målt OD mellom fotometerne.
Målt OD avhenger av cellenes form og størrelse
Celler i ulike prøver kan variere fra hverandre i cellestørrelse og cellefasong, og disse faktorene påvirker en prøve’s OD. Ifølge Koch 1970: “The standard curves on the same instrument [photometer] will not be the same for cultures of the same organism grown under different conditions where the cells have different shape or size distributions.” (Distribusjon refererer til at ikke alle cellene i en prøve har nøyaktig samme størrelse og fasong, det vil være litt variasjon mellom cellene innad i en prøve.)
Det Koch sier gjelder ikke bare for kulturer av samme organisme dyrket under ulike vekstforhold, men gjelder også for kulturer av ulike organismer dyrket under samme (eller ulike) vekstforhold.
I praksis betyr dette at dersom du har en standardkurve laget med et bestemt fotometer og en bestemt organisme dyrket under et bestemt vekstforhold, og deretter endrer enten fotometeret eller organismen eller vekstforholdet, så må du lage en ny standardkurve.
Del 4: Spørsmål og svar
Hvordan blir en absorbans- eller OD-kurve laget?
I seksjonen “Bølgelengde-velgeren” nevnte jeg at noen fotometre bruker lysfiltre for valg av bølgelengde, mens andre fotometre bruker et diffraksjonsgitter. For å sitere meg selv: Fordelen med et diffraksjonsgitter over et filter er at fotometeret kan rotere diffraksjonsgitteret for å justere hvilken bølgelengde som passerer gjennom utgangssprekken. Gitteret kan dermed velge en hvilken som helst bølgelengde innen et bestemt område, med små intervaller mellom hver valgbar bølgelengde.
La oss igjen se på absorbans-spekteret til o-nitrophenol (Figur 2 i denne teksten). Kurven viser absorbansen for o-nitrophenol for bølgelengder mellom 360 nm og 540 nm. Fotometre med diffraksjonsgitre brukes for å lage slike spektra. Alt en trenger å gjøre er å sette start-bølgelengden, slutt-bølgelengden, og intervallet i nm mellom hver målte bølgelengde. Fotometeret vil da automatisk måle absorbansen ved en rekke bølgelengder og generere et sett med datapunkter, som brukes til å lage absorbans-spekteret. (Denne metoden kan også brukes til å lage et transmittans-spekter eller et OD-spekter.)
(Siden er fotometer med et diffraksjonsgitter kan generere absorbans-spektra og andre typer spektra, så kalles et slikt fotometer gjerne et “spektrofotometer”. Filter-baserte fotometre, til tross for at de ikke kan lage spektra, har noen fordeler over spektrofotometre, inkludert lavere kostnad.)
Hvorfor måles absorbans ved bølgelengder som tilsvarer absorbans-topper?
Beer's lov sier at c × p × ε = log(1/T)
I denne ligningen blir ekstinksjonskoeffisienten ε behandlet som en konstant verdi. Men i virkeligheten har ε en variabel verdi. Dersom vi igjen ser på absorbans-spekteret til o-nitrophenol (Figur 2 i denne teksten), så ser vi at o-nitrophenol (ved høy pH) har en absorbans-topp ved 420 nm. Derfor absorberes lys ved 420 nm mer effektivt enn lys ved andre bølgelengder. Ettersom absorbans måles lengre og lengre unna 420 nm så blir absorbansen lavere og lavere og når eventuelt 0.
Dette betyr at for o-nitrophenol så er verdien for ekstinksjonskoeffisienten ε maksimal når fotometeret er stilt til 420 nm. Verdien for ε reduseres ettersom fotometeret stilles til bølgelengder lengre unna 420 nm. For bølgelengder langt nok unna 420 nm får vi ε = 0. Den viktige detaljen er at ε varierer med bølgelengden.
Husk at når fotometeret er satt til en bestemt bølgelengde, så treffes ikke prøven utelukket av lys av den bølgelengden. Istedet treffes prøven av et bølgelengde-bånd som er sentrert på den valgte bølgelengden. Dette betyr at prøven treffes av lys av ulike bølgelengder, og prøven vil absorbere noen av disse bølgelengdene mer effektivt enn andre. Dette kan, ihvertfall i teorien, forhindre en lineær relasjon mellom konsentrasjon og log(1/T). Dette vil videre forhindre en nøyaktig beregning av konsentrasjon ut ifra log(1/T).
Øverst på en absorbans-topp har absorbans-kurven en tangent med et stigningstall på 0. Altså, endring i absorbans som følge av endring i bølgelengde er minimal i området rundt absorbans-toppen. Dette betyr at ekstinksjonskoeffisienten ε har en tilnærmet konstant verdi i dette området. Derfor, når vi måler absorbans ved en bølgelengde som tilsvarer en absorbans-topp, og dersom båndbredden ikke er for stor, så vil Beer's lov gjelde og konsentrasjon har en lineær relasjon med log(1/T). (For å være nøyaktig: Beer's lov vil fremdeles ikke være 100% nøyaktig i disse tilfellene, den vil bare være en god tilnærming.)
Dersom vi istedet måler absorbans ved en av sidene til absorbans-toppen, eller dersom båndbredden er stor, så vil Beer's lov ikke gjelde (eller i det minste så blir det i teorien mindre lineæritet mellom konsentrasjon og log(1/T)).
Det er også en annen grunn til at absorbans typisk måles på en bølgelengde som korresponderer til toppen av absorbans-kurven, og denne grunnen er relatert til den første. Dette gjelder kun fotometre med diffraksjonsgitter, ikke fotometre med lysfilter. Når et fotometer er stilt til en gitt bølgelengde, si 420 nm, så skal det i teorien produsere et bølgelengde-bånd sentrert på 420 nm. Men dette skjer ikke alltid, ettersom et fotometer ikke kan sentrere båndet helt perfekt hver gang. Noen ganger kan båndet istedet være sentrert på for eksempel 419 nm eller 421 nm, et lite avvik fra verdien som båndet er ment å være sentrert på. Dette skjer fordi diffraksjonsgitteret ikke blir perfekt rotert hver gang, og det kan redusere reproduserbarheten av målingene. (Reproduserbarhet er fotometerets evne til å gi samme verdi av log(1/T) ved målinger av identiske prøver ved identiske fotometer-instillinger.)
Si at du setter bølgelengden til 420 nm, og fotometeret sentrerer båndet nøyaktig på 420 nm. Du tar en måling av en prøve o-nitrophenol med en gitt konsentrasjon. Senere setter du fotometeret til en annen bølgelengde for å måle noe annet. Og enda senere setter du fotometeret tilbake til 420 nm, og fotometeret sentrerer båndet ved 419 nm. Du tar igjen en måling av en prøve o-nitrophenol med samme konsentrasjon som tidligere.
Ettersom de to prøvene med o-nitrophenol er identiske, og ettersom fotometeret var satt til 420 nm hver gang, så skal du i teorien få nøyaktig samme verdi av log(1/T). Men fordi båndet var sentrert på 419 nm istedetfor 420 nm i den andre målingen, så vil log(1/T) fra den andre målingen ikke være identisk med log(1/T) fra den første målingen. Dette er likevel ikke noe problem: ettersom vi måler ved en bølgelengde som tilsvarer en absorbans-topp, så er det minimalt med endring i ekstinksjonskoeffisienten som følge av en liten endring i bølgelengden. De to målte verdiene av log(1/T) vil derfor i praksis være like.
Hvis vi måler absorbans ved en bølgelengde lengre unna absorbans-toppen, så vil en liten endring i båndets sentrering føre til en større endring i ekstinksjonskoeffisienten. Derfor vil målinger lengre unna absorbans-toppen gi mindre reproduserbare verdier av log(1/T).
(Alt i denne seksjonen har vært helt teoretisk: jeg har ikke sett noen data som støtter konklusjonen om at målinger ved bølgelengder som tilsvarer absorbans-topper gir bedre reproduserbarhet, eller bedre lineæritet mellom konsentrasjon og log(1/T). Men dersom noen har et fotometer tilgjengelig (for øyeblikket har ikke jeg det) så burde det være enkelt å teste denne teorien.)
Hvorfor blir OD typisk målt med 600 nm bølgelengde?
Se på Figur 5 in Myers 2013. Hver av de ni kurvene viser OD som en funksjon av bølgelengden brukt til å måle OD, og hver kurve representerer en bestemt organisme. Den blå prikkete kurven representerer OD av en E. coli-kultur som funksjon av bølgelengden. Kurven er ganske rett, og heller nedover i retning mot økt bølgelengde. Det samme stemmer også for den svarte kurven og den grå prikkete kurven, som representerer kulturer av mikroorganismene S. cerevisiae and A. vinlandii, selv om disse to kurvene ikke heller like bratt nedover som E. coli-kurven.
Myers skriver at “For an unpigmented culture such as E. coli, virtually any wavelength could be used [to measure the optical density], but [measuring] the OD at 600 nm has become a common convention – in part due to the ease of generating and measuring this wavelength within the visible spectrum.”
Ifølge denne teksten, “The use of 600 nm wavelength is based to some extent on a historical wavelength, when microbiologists used the simple Klett-Summerson photometer developed in 1939 and popular into the 1960s with fixed filters without having the possibility to adjust the wavelengths. The 600 nm filter had the advantage of providing low energy light that was not deleterious to the biologic sample and having low interference in most bacterial broth mixtures.”
(Forfatterne av teksten ovenfor bruker “600 nm filter” for å referere til et filter som absorberer lys utenfor 600 nm-området, som bare slipper gjennom bølgelengder nær 600 nm.)
Angående den første fordelen med 600 nm-filteret (“providing low-energy light”): såkalt høy-energi-lys (altså lys med kort bølgelengde, slik som UV-lys) kan forårsake DNA-mutasjoner som dreper celler. Dersom du skal fortsette å bruke en prøvekultur etter en fotometrisk måling, så vil du ikke at cellene skal ta skade. Lys med for eksempel 500 nm bølgelengde er ikke UV-lys, selv om det er sant at 500 nm-lys har mer energi enn lys ved lengre bølgelengder slik som 600 nm. Likevel så tror jeg ikke at kort eksponering til lys ved 500 nm (eller 400 nm for den saks skyld) vil skape noen betydelig celleskade, så jeg tror ikke dette er et godt argument for å bruke 600 nm som standard bølgelengde for måling av OD.
Angående den andre fordelen med 600 nm-lys (“low interference in most bacterial broth”): ifølge Myers 2013, “media containing cell or protein hydrolysate absorb light in the 350 to 550 nm region”. Lys med høyere bølgelengde absorberes derimot ikke av slike media. Resultatet er at slike media får en gul eller oransje farge.
I teorien, når du putter en blankprøve med rent medium i fotometeret og trykker på “zero”-knappen før du tar en måling, så vil fotometeret korrigere for redusert transmittans forårsaket av lysabsorpsjon i mediet. Husk det som ble skrevet tidligere:
log(1/Ts) – log(1/Tb)
= (ODkyvette,medium + ODceller) – (ODkyvette,medium)
= ODceller
Men, dersom bakteriene i kulturen vokser, så forbruker de næringsstoffer i mediet, slik at mediets evne til å absorbere lys endres over time. Korrigeringen log(1/Ts) – log(1/Tb) er derfor ikke en perfekt korrigering. En måte for å omgå dette problemet er å måle OD ved en bølgelengde hvor mediet ikke absorberer lys, for eksempel 600 nm (eller en hvilken som helst bølgelengde over 550 nm, ut i fra hva Myers sier).
(Hvis det skulle være nødvendig å måle OD i 350-550 nm-området, og bakteriene vokser i et medium som absorberer lys i dette området, så nevner Myers en metode som gir en mer nøyaktig korrigering: ikke bruk nytt medium som en blank. Istedet, ta ut to prøver fra den kulturen hvis OD skal måles. Den første prøven sentrifugeres for å separere cellene fra det flytende mediet (sentrifugeringen gjør at cellene danner en celleklump på bunnen av prøvebeholderen). Det sentrifugerte mediet, som nå er cellefritt, overføres til en kyvette og brukes som en blankprøve. Deretter kan den andre prøven måles (uten sentrifugering, slik at cellene fremdeles er suspendert i mediet). Med denne metoden vil mediet i blankprøven ha samme absorbans som mediet i bakterieprøven.)
Vi har nå forklaringen på hvorfor 600 nm er standard bølgelengde for måling av OD: før i tiden var det et mye brukt fotometer som kun hadde visse bølgelengder tilgjengelig, og en av disse bølgelengdene var bedre for å måle OD for kulturer hvor bakteriene vokser i et lys-absorberende medium. (Samt argumentet med at 600 nm lys har relativt lite energi, men som nevnt så er jeg noe skeptisk til gyldigheten av dette argumentet.)
La oss se på et tredje sitat omhandlende valg av bølgelengde, dette fra boken Brock Biology of Microorganisms: “Commonly used wavelengths for bacterial turbidity [OD] measurements include 480 nm, 540 nm, 600nm, and 660 nm. Sensitivity is best at shorter wavelengths, but measurements of dense cell cultures are more accurate at longer wavelengths.”
Dette er alt boken har å si om valg av bølgelengde. Tatt alene gir det ikke mye forståelse, men hvis vi igjen ser på Figur 5 i Myers 2013, på OD-kurven for E. coli, så kan siste setning fra sitatet blir bedre forstått. Ifølge Figur 5: for en gitt konsentrasjon av E. coli-celler, så vil måling av OD ved 400 nm gi en OD på ≈ 1.5, måling ved 500 nm gir ≈ 1.25, 600 nm gir 1.0, 700 nm gir ≈ 0.8, og 1000 nm (infrarød) gir ≈ 0.5. Forskjellen i OD er forårsaket av økt lysspredning ved kortere bølgelengder, og redusert lysspredning ved større bølgelengder.
Som følge av multiple scattering så har cellekonsentrasjon og OD kun en lineært relasjon opp til OD-verdier på rundt 0.5. For høyere cellekonsentrasjoner som gir OD-verdier over 0.5 vil relasjonen mellom OD og konsentrasjon ikke være lineær. Hvis vi istedet for å måle OD ved 600 nm velger en større bølgelengde, for eksempel 700 nm, så vil resultatet bli en reduksjon i OD for en gitt cellekonsentrasjon. (Dersom vi måler med 700 nm-lys, så vil dette skape en omtrent 20% reduksjon av OD relativt til dersom vi hadde målt ved 600 nm, når vi måler konsentrasjonen av E. coli-celler.) Som en konsekvens kan høyere cellekonsentrasjoner måles samtidig som at vi opprettholder den lineære relasjonen mellom OD og konsentrasjon. Dette er hva boken mener med “measurements of dense cell cultures are more accurate at longer wavelengths”.
Og angående bokens kommentar om sensitivitet: Hvis fotometeret, for en gitt cellekonsentrasjon, måler en relativt høy OD, så sies målingen å være sensitiv. Og hvis fotometeret, for en gitt cellekonsentrasjon, måler en relativt lav OD, så sies målingen å være insensitiv.
I Figur 5 i Myers 2013 ser vi at for en gitt cellekonsentrasjon så vil kortere bølgelengder gi høyere OD-verdier. Målinger utført ved kortere bølgelengder er altså mer sensitive enn målinger ved lengre bølgelengder.
“Sensitivitet” kan også brukes for å omtale fotometeret's evne til å skille mellom to prøver med lignende (men ikke identiske) konsentrasjoner. Med høyere sensitivitet kan mindre forskjeller mellom to prøver oppdages. Måling ved kortere bølgelengde gir høyere OD for en gitt cellekonsentrasjon. Dette vil resultere i en større absolutt OD-forskjell mellom to ulike cellekonsentrasjoner. Den større OD-forskjellen gjør det lettere å oppdage at de to cellekonsentrasjonene er ulike.
Jeg vet ikke om noen situasjon hvor bruk av kortere bølgelengde for å øke sensitivitet faktisk vil være nyttig. Spesielt når en tenker på at kortere bølgelengder reduserer konsentrasjons-området hvor OD og konsentrasjon har en lineær relasjon. Bruk av høyere bølgelengder for å opprettholde den lineære relasjonen ved høyere konsentrasjoner kan være praktisk, men du kan også opprettholde lineæriteten ved å redusere kyvettens path length, eller ved å fortynne prøvene. Personlig ville jeg generelt holdt meg til å måle OD-verdier ved 600 nm, kun fordi dette er vanligst.
(Angående bokens kommentar om at “Commonly used wavelengths for OD measurements include 480 nm, 540 nm, 600nm, and 660 nm”: jeg tror ikke noen av disse bølgelengdene unntatt 600 nm ofte brukes til å måle OD, eller ihvertfall så har jeg ikke sett dem brukt noe sted.)
Hvordan ble Beer's lov oppdaget?
Som du nå vet så sier Beer's lov at c × p × ε = log(1/T). Men hvor kommer denne ligningen fra? La meg si først som sist at jeg ikke kan gi et fullt svar, kun et delvis svar. Beer's lov kalles alternativt for “Bouguer-Lambert-Beer’s lov”. Dette navnet stammer fra tre vitenskapsmenn, hvis arbeider relevant for loven er:
1. Bouguer (1729): Essai d'optique sur la gradation de la lumière (engelsk oversettelse: Middleton (1961): Optical Treatise on The Gradation of Light)
2. Lambert (1760): Photometria sive de mensura et gradibus luminis, colorum et umbrae (engelsk oversettelse: DiLaura (2001): Photometria or On The Measure and Gradations of Light, Color, and Shade)
3. Beer (1852): Bestimmung der Absorption des rothen Lichts in farbigen Flüssigkeiten (Fri artikkel)
Jeg har ikke faktisk lest noen av disse (jeg nevner dem kun sånn at andre som eventuelt er interessert kan se på dem). De første to tekstene er bøker, mens den tredje teksten er en relativt kort artikkel som kun er tilgjengelig på tysk. Derfor, for å prøve å få en forståelse av Beer's lov må jeg bruke noen sekundære kilder.
Artikkelen Malinin 1961 inkluderer et oversatt utdrag fra Bouguer 1729. I utdraget skriver Bouguer at dersom et gjennomsiktig objekt deles opp i lag av lik tykkelse, så kan man anta at hvert lag vil fjerne (intercept) samme antall lysstråler. Dersom dette er tilfellet vil lysintensiteten reduseres "aritmetisk" for hvert lag. (Slik at dersom ett lag blokkerer 50% av lyset som går gjennom, så vil to lag blokkere 100% av lyset som går gjennom.) Bouguer fortsetter:
To examine the truth or the falsity of this idea, I passed a light of 32-candlepower perpendicularly through two sheets of glass, after which I found its intensity to have been halved, for it was then only 16 candlepower. Now if another thickness of two sheets of glass had caused an equal diminution, all the rays would have been intercepted. Nevertheless, the addition of two more sheets certainly did not form an absolutely opaque body. The light was still very bright, and when I passed it through ten sheets, it was still as intense as that from one candle.
La oss tenke på Bouguer's two sheets of glass som et "lag" av glass, og hans fire sheets som to lag av glass. Bouguer skriver at dersom det andre laget skulle ha blokkert samme antall lysstråler som det første laget, så måtte mengden lys som entrer det andre laget ha vært lik mengden lys som entrer det første laget. Men etersom det første laget allerede har blokkert halvparten av lysstrålene, så vil mengden lys som entrer det andre laget kun være halvparten av lyset som entrer det første laget. Dette betyr at det første laget blokkerer halvparten av det innfallende lyset, og det andre laget vil blokkere halvparten av lyset som transmitteres gjennom det første laget over til det andre. Dette reduserer lysintensiteten transmittert gjennom de to lagene til en kvart-del av intensiteten til det innfallende lyset. Intensiteten av lyset sies å reduseres "geometrisk" for hvert lag.
Bouguer formulerer ikke sine resultater med en matematisk formel, men det er lett å gjøre dette:
Is = I0 × (1/2)p
hvor I0 er intensiteten til det innfallende lyset på glasset, Is er intensiteten av lyset som kommer ut av glasset, og p er tykkelsen av glasset målt i “pairs of glass sheets”. Derfor, med et innfallende lys på “32 candlepower” og et lag glass har vi:
Is = 32 × (1/2)1 = 16
Med fire “sheets of glass” (to lag) har vi:
Is = 32 × (1/2)2 = 8
Og for ti “sheets of glass” (fem lag) har vi:
Is = 32 × (1/2)5 = 1
Formelen Is = I0 × (1/2)p kan generaliseres slik:
Is = I0 × (1/α)p
hvor I0 er intensiteten av lys som faller på et gjennomsiktig objekt, p er lysets "path length" gjennom objektet (path length måles i cm eller en annen valgt lengde-enhet), og Is er intensiteten av lyset som kommer ut av det gjennomsiktige objektet. 1/α er den andelen av innfallende lys som transmitteres gjennom en lengde-enhet av objektet. Denne andelen avhenger av objektet og er derfor ikke uttrykt som en bestemt verdi. Istedet så representerer α et tall større enn 1, slik at 1/α representerer en verdi mellom 0 og 1.
Ligningen ovenfor kan løses for p. Først deler vi begge sider av ligningen med I0. Da får vi:
Is/I0 = (1/α)p = 1p/ αp = 1/αp = α−p
logα(Is/I0) = logα(α−p) = −p
p = −logα(Is/I0) = logα(I0/Is)
Bruk logaritme-regelen for endring av basetall for å endre logα til log10 (som jeg skriver bare som log):
p = log(I0/Is) / log(α)
log(I0/Is) = p × log(α)
Husk fra tidligere i denne artikkelen at Is/I0 = Ts, slik at I0/Is = 1/Ts. Derfor har vi at:
log(1/Ts) = p × log(α)
Altså, logaritmen av resiprokalen av transmittansen er lik path length multiplisert med en koeffisient. Denne ligningen ligner på Beer's lov, bortsett fra at den ikke inkluderer konsentrasjonen “c” som en variabel.
Jeg har ikke klart å finne ut nøyaktig hva Lambert bidro med. Det var ihvertfall ikke han som inkluderte konsentrasjon som en variabel i ligningen. Da har vi igjen Beer. Var det han som la til konsentrasjon-variabelen? Nei, det var han faktisk ikke, eller ihvertfall ikke direkte. Pfeiffer 1951 gir noen detaljer om Beer's 1852-artikkel, men til tross for disse detaljene så forstår jeg ikke helt hva det var Beer drev med. Men, denne paragrafen fra siste side av Pfeiffer er interessant:
It is accordingly not surprising to find in Beer's paper no evidence that he believed himself to have discovered a new absorption law. In a thorough (but necessarily incomplete) investigation of the older literature, we found the phrase "Beer's Law" first used by Walter nearly forty years [after Beer released his article]. Kayser, in his monumental "Handbuch der Spectroscopie", says:
“If we let a represent the absorption coefficient for unit concentration, there follows the absorption law I = I0 × adc, where d is the thickness of the layer and c is the concentration. This law was set up by Beer and is called Beer's Law.”
Comparison of equation (11) [the equation in Kayser’s "Handbuch der Spectroscopie"] with equation (1) [Beer’s equation] shows the first half of the last sentence to be incorrect [that is, Beer didn’t set up any kind of law]. There is thus good precedent for any twentieth-century inaccuracies in describing what Beer thought and did.
Selv om jeg ikke helt forstår hva Beer drev med, og selv om jeg ikke kan lese tysk, så er det veldig lett å se gjennom Beer’s artikkel og sjekke at noe slikt som I = I0 × adc ikke står noe sted i Beer's tekst. Så hvem vet hvorfor denne Kayser-typen påsto at Beer formulerte denne ligningen når det er klart at han ikke gjorde noe slikt.
Kilder
Koch
(1970): Turbidity Measurements of Bacterial Cultures in Some Available Commercial Instruments. Betalt artikkel
Madigan, Martinko, Bender, Buckley, Stahl
(2015): Brock Biology of Microorganisms, 14th edition. [Book]
Malinin, Yoe
(1961): Development of the Laws of Colorimetry. Betalt artikkel
Monod
(1949): The growth of bacterial cultures. Fri artikkel
Myers, Curtis, Curtis
(2013): Improving accuracy of cell and chromophore concentration measurements using optical density. Fri artikkel
Pfeiffer, Liebhafsky
(1951): The Origins of Beer's law. Betalt artikkel