Selenocystein-innsettingssekvensen i E. coli
© Anders Skovly 2025
Som beskrevet i artikkelen om oversetting så blir aminosyren selenocystein satt inn i proteiner via en unik mekanisme som ikke deles av noen av de andre aminosyrene. I denne artikkelen presenteres utvalgte metoder og resultater fra syv forskningstekster omhandlende denne mekanismen. Dette er et nisje-tema og er vel hovedsakelig av interesse for de som skal genmodifisere et gen med en selenocystein-kodon. Artikkelen kan likevel være av interesse for andre, ettersom den beskriver diverse molekylære teknikker.
Zinoni, Heider, Bock (1990): Features of the formate dehydrogenase mRNA necessary for decoding of the UGA codon as selenocysteine
De aller fleste UGA-kodoner fungerer som stopp-kodoner, men noen få UGA-kodoner fungerer istedet som selenocystein-kodoner. Altså, disse UGA binder ikke til RF2 (release factor 2), men til tRNASecACU (antikodonet er skrevet fra 3'-enden mot 5'-enden). Et eksempel er E. coli’s fdhF-gen, hvor kodon nummer 140 er en UGA som oversettes til selenocystein.
Binding av tRNASecACU til UGA avhenger av tilstedeværelsen av et protein kalt SelB. Dette til forskjell fra alle andre elongator-tRNA, som avhenger av proteinet EF-Tu (Elongeringsfaktor Tu) for å binde til sine kodoner. SelB og EF-Tu har stor likhet i aminosyresekvens, men SelB inneholder en "ekstra del" (ekstra aminosyresekvens) på den ene enden av kjeden som ikke finnes hos EF-Tu. I 1990 var det uvisst hvilken funksjon denne ekstra delen hadde.
Zinoni antok at noen mRNA, slik som de transkriptert fra fdhF-genet, inneholder en bestemt struktur hvis funksjon er å fasilitere binding av tRNASec til UGA. Han tenkte at denne strukturen kunne være en eller flere stamme-løkker. (En stamme-løkke er en region i RNA hvor to sekvenser baseparer til hverandre og danner en dobbelheliks kalt en stamme, mens en tredje sekvens, kalt en løkke, forbinder de to baseparede sekvensene.) mRNA fra fdhF-genet ble derfor analysert for mulige stamme-løkker nær UGA, og tre mulige strukturer ble identifisert (se Zinoni’s Figur 1).
Første forsøk: metode
Zinoni laget ni fdhF deletion-mutanter hvor tilfeldige lengder av fdhF-genets 3’-ende ble fjernet. Dette for å undersøke hvor stor del av 3’-enden som kan fjernes før UGA-oversettelse reduseres, og hvor stor del som kan fjernes før UGA-oversettelse elimineres fullstendig. Data fra en slik undersøkelse kan brukes til å vurdere om noen av de tre identifiserte stamme-løkkene i mRNA virkelig er involvert i UGA-oversettelse.
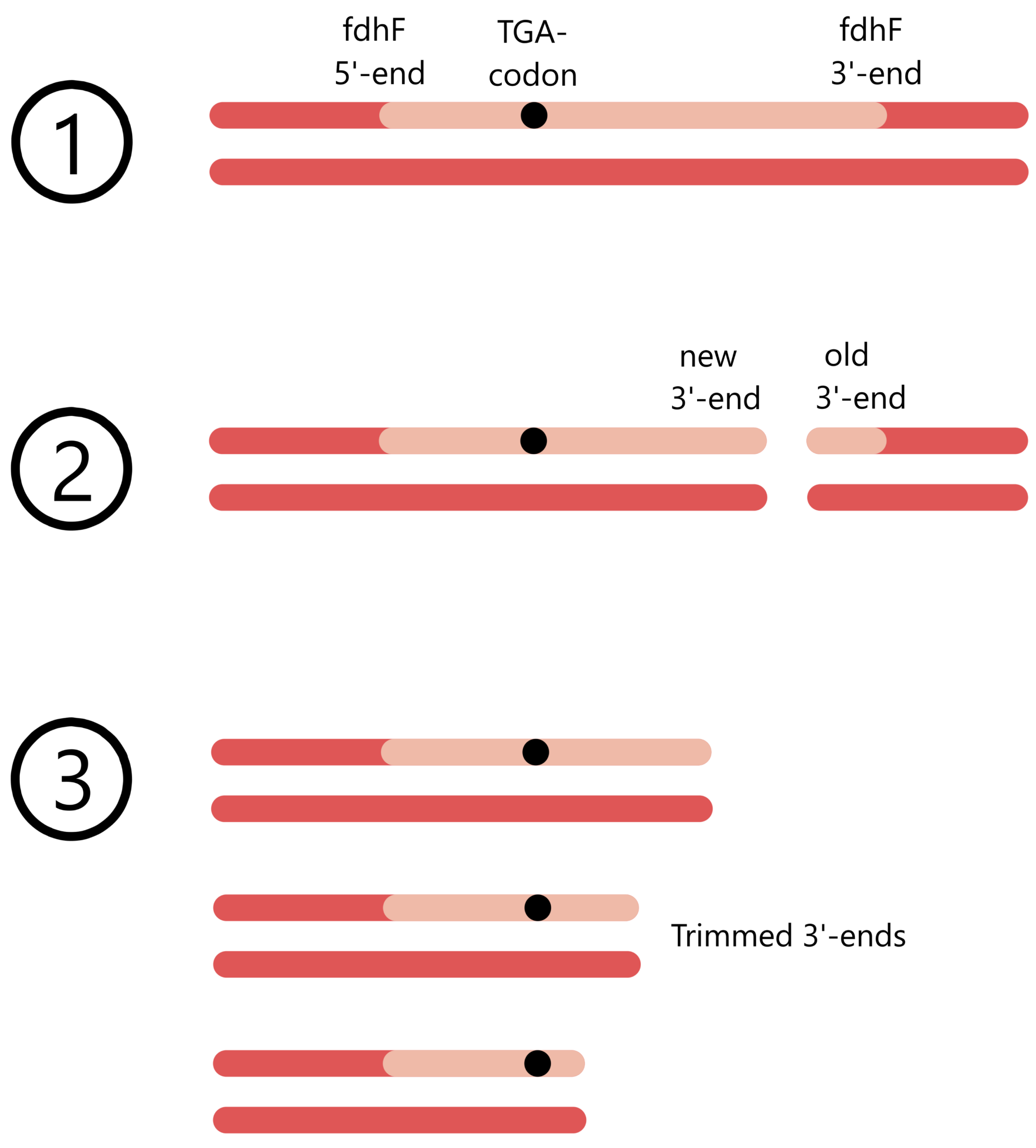
I en mutant var UGA-kodonet etterfulgt av fire baser nedstrøms, mens resten av 3’-enden var fjernet. Denne mutanten vil refereres til som Mut-4. I de andre mutantene var UGA etterfulgt av henholdsvis 7, 18, 27, 39, 46, 47, 75, og 384 baser. Disse vil refereres til som Mut-7, Mut-18, etc. (Bare for informasjon, det komplette fdhF-genet inneholder 1728 baser nedstrøms av UGA.)
Graden av UGA-oversettelse i fdhF deletion-mutantene kan ikke måles direkte, men kan måles indirekte ved først å lage fdhF-lacZ-fusjonsgener, sette fusjonsgenene inn i E. coli-celler, og deretter måle noe kalt β-galaktosidase-aktivitet.
Et fdhF-lacZ-fusjonsgen lages ved å sette inn en fdhF-mutant like ved starten (5’-enden) av et lacZ-gen. E. coli vil transkriptere fusjonsgenet fra fdhF-promoteren til lac-terminatoren, slik at det dannes fusjonerte mRNA. Disse mRNA vil oversettes fra fdhF's start-kodon til lacZ's stopp-kodon, og oversettes derfor til et fusjonsprotein. Hver av de ni fdhF-mutantene settes inn i hvert sitt fusjonsgen, og disse ni fusjonsgenene settes inn i hver sin E. coli-kultur, slik at β-galaktosidase-aktivitet kan måles separat for hver mutant.
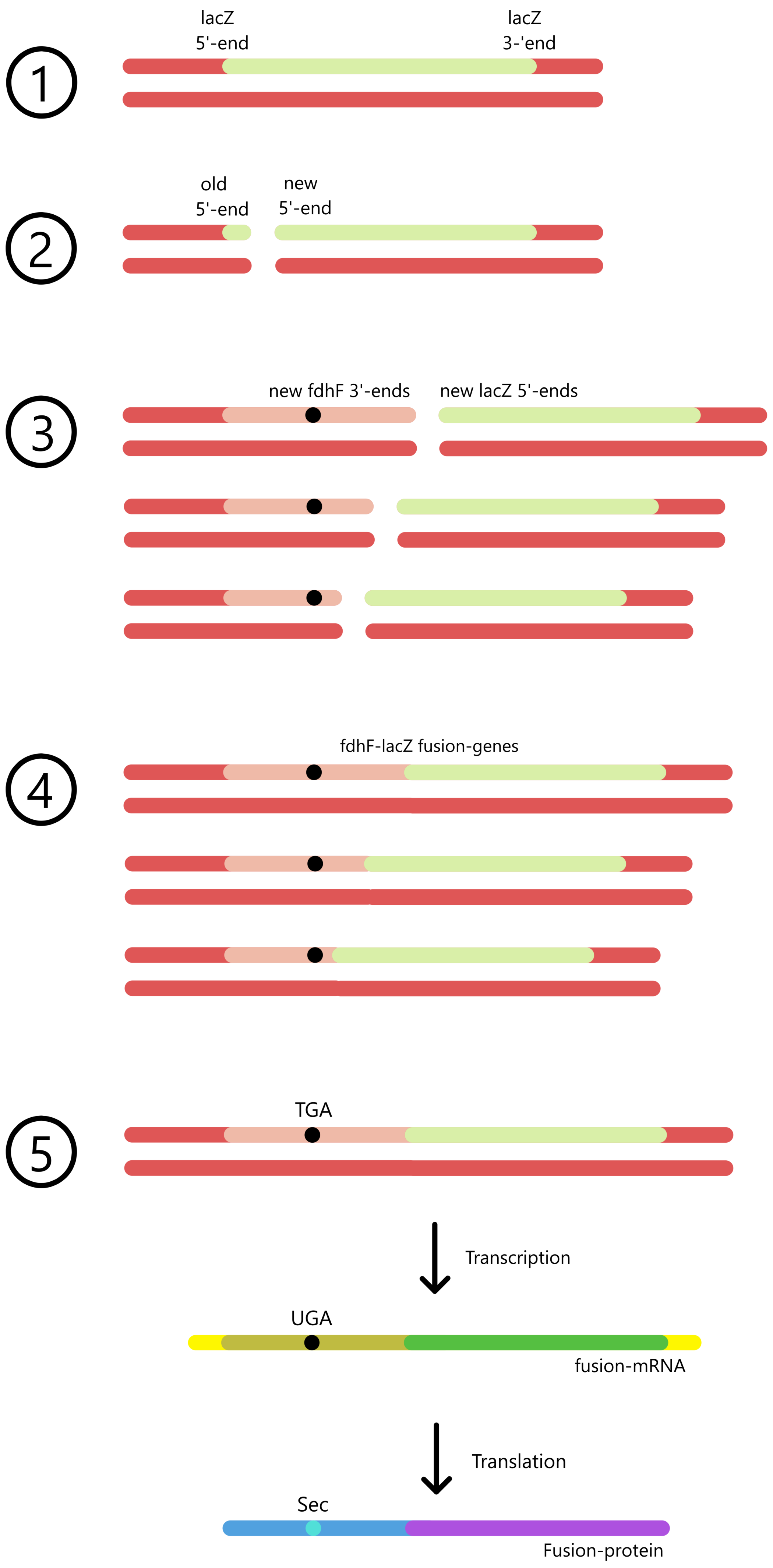
(lacZ-genet i E. coli's kromosom etterfølges nedstrøms av lacY- og lacA-genene, hvor lac-terminatoren først kommer etter lacA. I dette eksperimentet benyttes et lacZ-gen i en plasmid, denne lacZ etterfølges direkte av en terminator, uten andre gener imellom lacZ og terminatoren.)
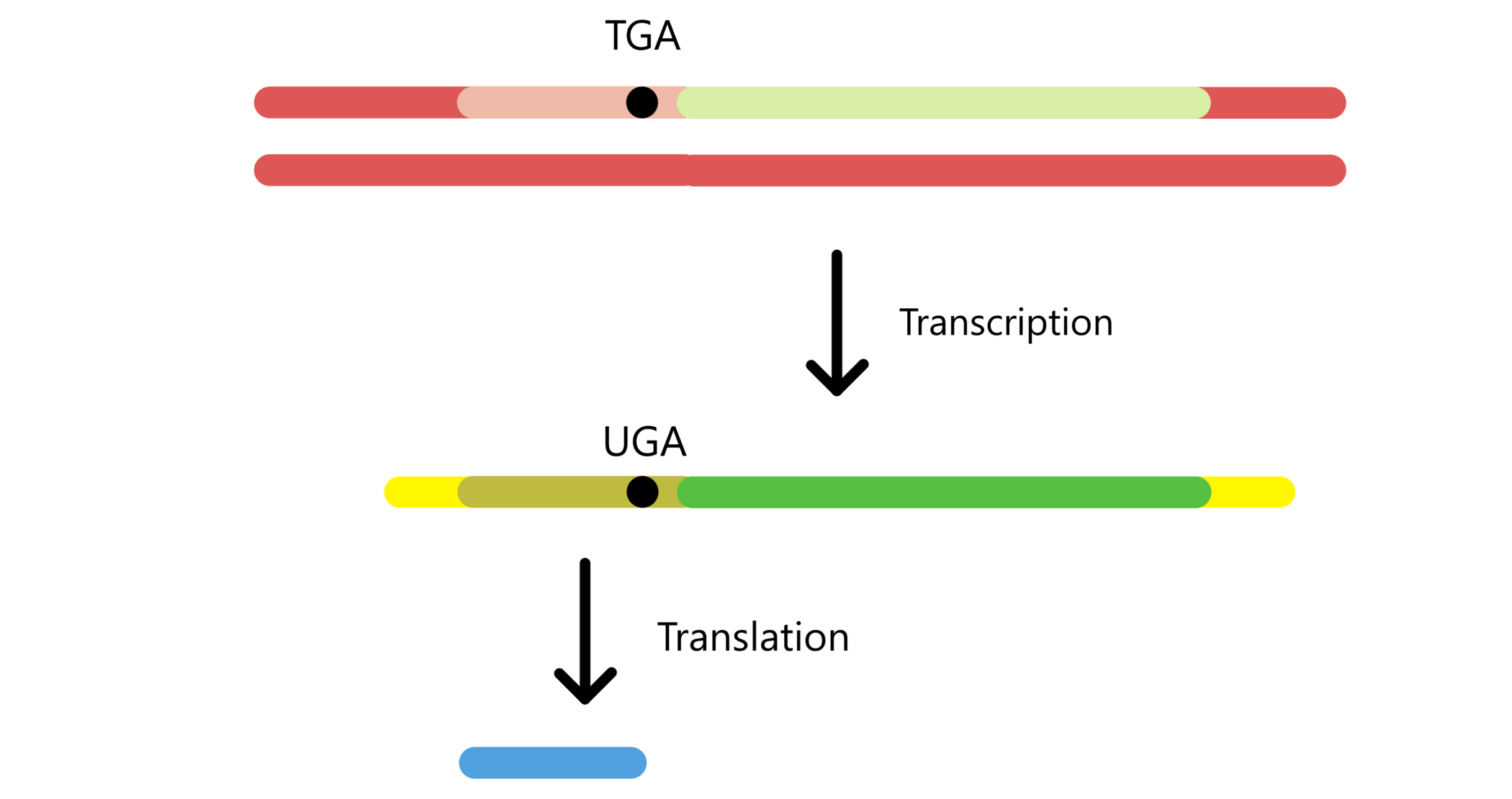
Ettersom fdhF ble satt inn 5’ for lacZ-genet, og fordi protein-meldinger oversettes fra 5’-enden mot 3’-enden, så oversettes fdhF-delen av meldingen først og lacZ-delen av meldingen sist. Dette betyr at oversetting av lacZ avhenger av oversetting av fdhF. Hvis fdhF’s UGA-kodon binder til tRNASecACU og oversettes til selenocystein vil også lacZ oversettes, og det produseres et fusjonsprotein. Alternativt kan UGA binde til RF2 (Release Factor 2) og fungere som stopp-kodon, og i såfall produseres det ikke noe fusjonsprotein.
Antallet fusjonsproteiner i en E. coli-kultur kan kvantifiseres indirekte ved å bestemme β-galaktosidase-aktiviteten i kulturen. lacZ er genet for β-galaktosidase, et protein som bryter ned β-galaktosidaser (for eksempel laktose, som kan brytes ned til galaktose og glukose). Et fdhF-lacZ-fusjonsprotein vil også ha denne evnen til å bryte ned β-galaktosidaser. β-galaktosidase-aktivitet er et mål på hvor raskt β-galaktosider brytes ned og er proporsjonalt med antallet fusjonsproteiner.
For å bestemme β-galaktosidase-aktivitet benyttes en menneskeskapt β-galaktosid kalt ONPG (o-nitrophenyl-β-galaktose), som kan brytes ned til galaktose og o-nitrophenol. ONPG er fargeløst og absorberer ikke lys, mens o-nitrophenol absorberer lys rundt 420 nanometers bølgelengde og er derfor gulfarget.
Kort forklart så bestemmes β-galaktosidase-aktivitet ved å tilsette ONPG til E. coli-kulturer, vente en bestemt tid for å la β-galaktosidasene splitte ONPG, for deretter å måle hvor mye lys som absorberes av kulturene. Absorbsjonen av lys måles med et spektrofotometer stilt til å bruke lys av 420 nm bølgelengde. Spektrofotometeret gir resultatene i form av absorbans-verdier, disse brukes til å beregne Miller-verdier som representerer β-galaktosidase-aktiviteten i kulturene. (Lengre ned i denne artikkelen finnes en seksjon som utdyper om hvordan Miller-verdiene beregnes.)
Altså: en Miller-verdi er proporsjonale med konsentrasjonen av o-nitrophenol i en kultur, som er proporsjonal med konsentrasjonen av fdhF-lacZ-fusjonsproteiner, som er proporsjonal med hvor effektivt UGA oversettes til selenocystein.
Første forsøk: resultater
De beregnede Miller-verdiene for de ni ulike fdhF-variantene er presentert i Zinoni’s Figur 2A. Mut-4, Mut-7, og Mut-18 viser ingen UGA-oversettelse (Miller-verdier ≈ 0), mens Mut-27 og Mut-39 viser ineffektiv UGA-oversettelse (Miller-verdier ≈ 200 og 700). Mut-46 har en Miller-verdi på ≈ 1500, denne verdien opprettholdes i Mut-47 og Mut-75.
Det antydes derfor at Mut-46/47/75 kan danne den komplette mRNA-strukturen nødvendig for maksimal UGA-oversettelse, mens Mut-27/39 kan danne ukomplette/ustabile strukturer som fasiliterer en lavere grad av UGA-oversettelse.
I Mut-384 stiger Miller-verdien til ≈ 2200. Ettersom Miller-verdien er konstant ≈ 1500 fra Mut-46 til Mut-75 så antar Zinoni at forskjellen i Miller-verdi mellom Mut-46/47/75 og Mut-384 skyldes "translational reinitiation events", altså at Mut-384 ikke faktisk har høyere UGA-oversettelse enn Mut-46/47/75 (jeg vet ikke hvordan "translational reinitiation" fungerer, så kan ikke kommentere på dette).
Av de tre mRNA-strukturene som ble presentert i Zinoni's Figur 1 så er det struktur 1C som støttes best av resultatet. Mut-39, som viser redusert UGA-oversettelse, kan ikke danne det nederste AU-paret i 1C-stammen, mens Mut-46, som viser maksimal UGA-oversettelse, kan danne alle av 1C-stammens basepar. (Jeg synes likevel at det virker rart at kun ét ekstra AU-par i stammen kan føre til mer enn en dobling av UGA-oversettelse, fra 700 Miller i Mut-39 til 1500 Miller i Mut-46.)
(Dersom en ser på Zinoni’s Figur 2A så er det en mutant «−7» med en Miller-verdi på ≈ 6000, mye høyere enn i de andre mutantene. Denne mutanten nevnes ikke i teksten, men må være en tiende mutant hvor UGA-kodonet ble fjernet fra fdhF sammen med syv baser oppstrøms av UGA (samt alle basene nedstrøms av UGA). At Miller-verdien øker så mye når UGA-kodonet fjernes hinter om at UGA-oversettelse generelt er lite effektivt sammenlignet med oversetting av andre kodoner.)
Andre forsøk
For å verifisere resultatet fra forrige forsøk ble et nytt forsøk utført med en alternativ metode. Først ble E. coli med de ni mutantene dyrket i medium tilsatt radioaktiv selenium, slik at bakteriene produserer radioaktiv selenocystein. Når bakteriene så danner proteiner med selenocystein blir proteinene også radioaktive. Dersom bakteriene danner radioaktive fdhF-lacZ-fusjonsproteiner vil dette bety at de fusjonerte mRNA kan danne strukturen nødvendig for UGA-oversettelse.
Proteinene i E. coli ble ekstrahert, separert fra hverandre med gelelektroforese, og analysert med autoradiografi (som gjør det mulig å se tilstedeværelse av radioaktivt materiale). E. coli med Mut-4, Mut-7 og Mut-18 hadde ingen radioaktive fusjonsproteiner, E. coli med Mut-28 hadde en liten mengde, de med Mut-39 hadde en moderat mengde, og de med Mut-46, Mut-47, Mut-75, og Mut-384 hadde en stor mengde (se Zinoni’s Figur 3). Dette resultatet stemmer overens med Miller-verdiene fra forrige forsøk.
Siste forsøk
I et siste forsøk ble det laget fem fdhF deletion-mutanter hvor varierende lengder av genets 5'-ende ble fjernet, for å undersøke hvor stor del av fdhF-genets 5’-ende som kan fjernes før UGA-oversettelse reduseres. I en mutant var UGA-kodonet forbigått av (preceded by) én base mens resten av 5'-enden var fjernet, denne vil refereres til som Mut-1(5'). I de andre mutantene var UGA forbigått av henholdsvis 9, 18, 42, og 67 baser, disse vil refereres til som Mut-9(5'), Mut-18(5'), etc.
Mutantene ble brukt til å lage et nytt sett fdhF-lacZ-fusjonsgener. Som i forrige forsøk ble disse satt inn i E. coli-celler, og β-galaktosidase-aktivitet samt mengde radioaktive proteiner ble testet. Resultatet er vist i Zinoni's Tabell 2 og Figur 4, og viser at UGA-oversettelse fungerer i alle fem mutantene. Mut-1(5') har den laveste β-galaktosidase-aktiviteten på 1758 Miller (i E. coli dyrket med glycerol), mens Mut-9(5') har en betydelig høyere verdi på 4908 Miller. I Mut-18(5') og Mut-42(5') går verdiene noe ned, til 3919 og 4406 Miller, mens i Mut-67(5') går de opp til 6144 Miller.
Det at β-galaktosidase-aktiviteten stiger så mye fra Mut-1(5') til Mut-9(5') antyder at de første ni basene 5' for UGA er involvert i UGA-oversettelse. Dette er kompatibelt med mRNA-struktur 1A og 1B, men ikke med struktur 1C hvor stamme-og-løkken ikke involverer noen base 5' for UGA. At β-galaktosidase-aktiviteten reduseres fra Mut-9(5') til Mut-18(5') og Mut-42(5') er et rart resultat, det er ingen mulig forklaring på dette. Denne reduksjonen er ikke kompatibelt med mRNA-struktur 1A, hvor atten baser foran UGA er nødvendig for å danne den ene stamme-løkken, mens førti baser foran UGA er nødvendig for å danne hele 1A-strukturen.
Resultatet fra første forsøk passer altså best med mRNA-struktur 1C, mens resultatet fra siste forsøk passer best med 1B. Zinoni foreslår en mulig mekanisme hvor mRNA først inntar struktur 1B, og at når ribosomet nærmer seg UGA og bryter baseparene 5' for UGA så vil mRNA innta struktur 1C. Med en slik mekanisme kan begge strukturene være involvert i UGA-oversettelse.
(Miller-verdiene fra siste forsøk er generelt mye høyere enn verdiene i første forsøk. Jeg antar dette er fordi i første forsøk benyttes fdhF-promoteren og fdhF's start-kodon i transkripsjon og oversetting av fusjonsgenene, mens i siste forsøk benyttes lac-promoteren og lacZ's start-kodon. Ulike promotere og start-kodoner kan gi ganske ulik grad av transkripsjon og oversetting, noe som vil påvirke konsentrasjonen av fusjonsproteiner. Miller-verdiene fra de to forsøkene kan derfor ikke sammenlignes mot hverandre.)
(Lengre ned i denne artikkelen finnes en seksjon som utdyper om Zinoni's forsøksmetode.)
Heider, Bock (1992-a): Targeted Insertion of Selenocysteine into the α Subunit of Formate Dehydrogenase from Methanobacterium formicicum
Bakterien Methanobacterium formicicum har et gen kalt fdhA. Dette genet inneholder ikke noen selenocystein-kodoner, og følgelig blir ikke genet oversatt til proteiner med selenocystein. Heider forsøkte å mutere fdhA-genet slik at det får en UGA-kodon som oversettes til selenocystein. Dette ble gjort ved å endre en av genets UGC-kodoner til et UGA-kodon, samtidig som at det nær denne UGA ble innført mutasjoner som fasiliterer dannelsen av stamme-løkker lignende de i Zinoni 1990 Figur 1.
De muterte fdhA-genene ble satt inn i E. coli-celler, og grad av UGA-oversettelse ble testet med radioaktiv selenium-metoden (samme metode som ble brukt av Zinoni). Ved å se hvilke mutasjoner som skaper UGA-oversettelse og hvilke mutasjoner som ikke gjør det kan man få en bedre forståelse av mRNA-strukturen som kreves for UGA-oversettelse.
Først ble det laget to mutanter kalt U2 og D2. Mutant U2 kan danne en stammeløkke hvor UGA-kodonet er posisjonert i løkken. Denne stammeløkken ligner de som ble vist i Zinoni's figur 1A og 1B (se Heider's figur 1, stammeløkken merket "E. coli" er den lange stammeløkken fra Zinoni's figur 1A og kan også danne den korte stammeløkken fra Zinoni's figur 1B). Mutant D2 kan danne en stamme lignende den i Zinoni's figur 1C (se Heider’s figur 2, stammeløkken merket "E. coli" er stammeløkken fra Zinoni's figur 1C).
I Heider's Figur 5 ser vi at det ikke er UGA-oversettelse i verken av de to fdhA-mutantene (proteiner fra mutant U2 er i kolonne 6 og proteiner fra mutant D2 er i kolonne 8, verken av kolonnene inneholder et protein med radioaktiv selenocystein).
Løkken i fdhA mutant U2 er ganske ulik den tilsvarende løkken i E. coli's fdhF (se Heider's Figur 2). I tillegg har stammenløkken i E. coli en uracil som stikker ut fra stammen nær løkken, denne utstikkende uracil mangler i mutant U2. Det ble derfor laget en ny fdhA-mutant D22 hvor den apikale delen av stammeløkken i mutant D2 var erstattet av tilsvarende del fra E. coli (se Heider's Figur 2).
I Heider's Figur 5 ser vi at fdhA-mutant D22 fasiliterer UGA-oversettelse (proteiner fra mutant D22 er i kolonne 14, på stedet markert 93 kDa. Jeg vet ikke hva den svake stripen under 80 kDa representerer).
Mutasjonene i U2 og i D22 ble kombinert i en og samme mutant U2D22. Dette for å teste om kombinasjonen av U2- og D22-mutasjonene gir bedre UGA-oversettelse enn D22-mutasjonene alene. Dette var ikke tilfellet: U2D22 viste en lignende grad av UGA-oversettelse (lignende mengde radioaktive proteiner) som mutant D22 (se Heider’s Figur 5, proteiner fra mutant U2D22 er i kolonne 15).
I Zinoni 1990 ble det foreslått at stammeløkkene i 1B og 1C (fra Zinoni’s Figur 1) begge er involvert i UGA-oversettelse, ved at fdhF-mRNA først inntar stammeløkkene i 1B, og når ribosomet nærmer seg UGA så inntar mRNA stammeløkken i 1C. Heider fant at fdhA-mutant D22 har UGA-oversettelse, noe som støtter ideen om at stammeløkke 1C er involvert i UGA-oversettelse. Heider's resultat gir dog ingen støtte til at stammeløkke 1B bidrar i oversettelsen: fdhA-mutant U2 viste ingen UGA-oversettelse, og den kombinerte mutanten U2D22 viste ikke betydelig høyere grad av UGA-oversettelse enn mutant D22.
Heider, Baron, Bock (1992-b): Coding from a distance: dissection of the mRNA determinants required for the incorporation of selenocysteine into protein
På dette punktet har det ikke blitt bevist at stammeløkken i Zinoni 1990 Figur 1C er den mRNA-strukturen som fasiliterer UGA-oversettelse, men dette er den hypotesen som er best støttet av resultatene til Zinoni og Heider. Herifra vil basesekvensen som danner stammeløkken i Figur 1C refereres til som "SECIS" (selenocysteine insertion sequence), og selve stammeløkken vil refereres til som "SECIS-stammeløkken".
For å undersøke hvordan UGA-oversettelse blir påvirket av mutasjoner i SECIS-løkken laget Heider seksten fdhF-mutanter, hver med en av SECIS-løkkens seks baser endre til en annen base (se Heider’s Figur 3). UGA-oversettelse i de seksten mutantene ble testet ved å lage fdhF-lacZ-fusjonsgener og måle fusjonsproteinenes β-galaktosidase-aktivitet, samt ved å se hvor mange radioaktive fusjonsproteiner som lages. Et fusjonsgen som inneholdt en villtype (ikke-mutert) SECIS ble brukt som referanse for effektiv UGA-oversettelse.
Resultatet fra aktivitetstesten var at de fleste mutantene bare hadde noen få prosents UGA-oversettelse sammenlignet med villtypen (WT) (se Heider’s Figur 4A). Resultatet fra radioaktivitetstesten samsvarte med dette: mutantene har lite eller ingen synlig produksjon av fusjonsproteiner sammenlignet med villtypen (Heider's Figur 4B, fusjonsproteinene er på posisjonen merket "120 kDa"). Altså ser det ut til at alle basene i SECIS-løkken viktige for UGA-oversettelse.
(De to hypotetiske løkke-variantene AGAUCU og AGGCCU ble ikke testet. Hver av sekvensene er en såkalt revers komplement, så jeg antar dette er grunnen til at Heider ikke tested dem (han gir ikke selv noen kommentar til grunnen). Personlig ville jeg ha testet disse to sekvensene sammen med de andre seksten, bare for å ha testet hver eneste mulige base-mutasjon.)
I et annet forsøk ble det undersøkt om SECIS-stammens lengde påvirker UGA-oversettelse. To mutanter ble produsert: en med tre ekstra basepar satt inn i bunnen av stammen, og en med tre basepar fjernet fra bunnen av stammen (se Heider’s Figur 6B). (Merk at innsetting eller fjerning av tre basepar ikke påvirker oversettingen av kodoner, fordi kodonene forblir i samme leseramme. Leserammer forklares i seksjonen om Zinoni's forsøksmetode.)
Mutanten med tre ekstra basepar hadde 50% redusert β-galaktosidase-aktivitet sammenlignet med villtypen, mens mutanten med tre færre basepar hadde 100% reduksjon (se Heider’s Figur 6C). Det kan derfor se ut som at lengden på SECIS-stammen i villtypen både er en minimumslengde og en optimal lengde.
Som skrevet i seksjonen om Zinoni 1990: Binding av tRNASecACU til UGA avhenger av tilstedeværelsen av et protein kalt SelB. Dette til forskjell fra alle andre elongator-tRNA, som avhenger av proteinet EF-Tu for å binde til sine kodoner. SelB og EF-Tu har stor likhet i aminosyresekvens, men SelB inneholder en "ekstra del" (ekstra aminosyresekvens) på den ene enden som ikke finnes hos EF-Tu.
Heider foreslår at den ekstra aminosyresekvensen i SelB muligens danner en struktur som kan binde til de seks basene i SECIS-løkken, og at SECIS-stammens funksjon er å holde løkken (og dermed SelB + tRNASecACU) i riktig posisjon i forhold til ribosomet (se Heider’s Figur 9). Når UGA entrer ribosomets A-sted vil tRNASecACU da være posisjonert like ved A-stedet og kan binde til UGA før RF2 (release factor 2) kan gjøre det. UGA kan dermed fungere som selenocystein-kodon istedet for stopp-kodon.
(Lengre ned i denne artikkelen finnes en seksjon som utdyper om Heider's forsøksmetode.)
Baron, Heider, Bock (1993): Interaction of translation factor SELB with the formate dehydrogenase H selenopolypeptide mRNA
[In vivo-forsøk vs in vitro"-forsøk: Typisk så ønsker man å finne ut hvordan molekyler interagerer med hverandre inne i cellen (altså i molekylenes naturlige kjemiske og molekylære miljø). Forsøk som undersøker dette kalles in vivo (i liv)-forsøk. All forskning som presenteres i denne artikkelen er basert på in vivo-forsøk, med unntak av Baron 1993, som er fokuset for denne seksjonen. Visse analyser kan ikke utføres på molekyler inne i celler, og av denne grunn benytter Baron celle-frie metoder hvor noen få molekyler isoleres og studeres utenfor cellen. Slike forsøk kalles in vitro (i glass)-forsøk. ("Glass" refererer her til glassflasker og glassrør som brukes i laboratoriet når man studerer molekyler. Det er ikke et helt ideelt navn, ettersom celler i in vivo-forsøk også vil holdes i glassbeholdere.) Det molekylære miljøet kan påvirke hvordan molekyler interagerer sammen, så en må huske at resultater fra in vitro-forsøk ikke alltid er representative for hvordan molekyler interagerer inne i cellen.]
Heider foreslo at SelB, i tillegg til å binde til tRNASecACU og ribosomet, også kan binde til SECIS-løkken i fdhF-mRNA. Baron undersøkte om SelB kan binde til en RNA som inneholdt SECIS ("SECIS-RNA") ved bruk av metoden "electrophoretic mobility shift assay". Denne metoden er basert på gelelektroforese, altså bruk av et elektrisk felt til å bevege molekyler gjennom en gele. Molekylers bevegelseshastighet avhenger av deres størrelse, fasong, og elektrisk ladning. Dersom to molekyler binder sammen, for eksempel et protein og en SECIS-RNA, så kan dette endre deres hastighet gjennom geleen. En slik endring refereres til som et "mobilitetsskift", derav metodens navn.
Prøver med SECIS-RNA ble tilsatt varierende konsentrasjoner av SelB. SECIS-RNA var på forhånd gjort radioaktivt slik at dets posisjon i geleen kunne ses via autoradiografi. Resultatet var at tilsetting av store konsentrasjoner SelB ("10x/25x molar excess") forårsaker mobilitetsskift. Altså indikeres binding mellom SECIS-RNA og SelB (se Baron’s Figur 1A). Med den høyeste konsentrasjonen av SelB (25x molar excess) er det dog en god andel SECIS-RNA som ikke beveger seg gjennom gelen i det heletatt, og istedet bare blir værende på startstedet.
Som nevnt før så ligner aminosyresekvensen i SelB på sekvensen i EF-Tu, og det var kjent at EF-Tu avhenger av binding til GTP (guanosin trifosfat) for å fungere i oversettingsprosessen. Det ble derfor utført et andre forsøk hvor SECIS-RNA og SelB var blandet med GTP. I tillegg ble et tredje forsøk utført hvor GTP var erstattet av det strukturelt lignende ATP (adenosin trifosfat).
Tilsetting av ATP førte ikke til en betydelig endring av SECIS-RNA's bevegelse gjennom gelen. Tilsetting av GTP førte derimot til at all SECIS-RNA beveget seg bort fra startstedet (se Baron’s Figur 1A), og Baron kommenterer at GTP øker gele-løseligheten til RNA/SelB-komplekset. (Jeg antar dette betyr at SelB, i fravær av GTP, har en tendens til å klumpe seg sammen med andre SelB, og at klumpingen reduserer bundet SECIS-RNA's evne til å bevege seg gjennom gelen.) Dette beviser ikke at SelB er avhengig av binding til GTP for å fungere i UGA-oversettelse, men det viser at GTP har en interaksjon med RNA/SelB-komplekset.
Binding av et protein, slik som SelB, til en RNA kan være enten spesifikk (binding til en spesifikk RNA-sekvens/struktur) eller uspesifikk (binding til en hvilken som helst RNA-sekvens). For å teste om SelB binder spesifikt til RNA som inneholder SECIS ble prøver med SelB blandet både med SECIS-RNA og med diverse annet RNA, inkludert blant annet 5S rRNA. Dersom SelB kan binde til en hvilken som helst RNA-sekvens vil en del av SelB-proteinene binde de andre RNA, slik at mindre SelB blir tilgjengelig for binding til SECIS-RNA. Følgelig vil en mindre mengde SECIS-RNA bli mobilitetsskiftet.
Resultatet fra disse prøvene er ikke presentert i noen figur, men Baron kommenterer at det ikke var noen reduksjon i mobilitetsskiftet SECIS-RNA. Dette indikerer at SelB binder spesifikt til SECIS-delen i RNA.
Mobility shift assay ble også brukt til å undersøke binding mellom SelB og en RNA som inneholdt en SECIS-mutant hvor tredje base i løkken var endret fra guanin til cytosin. I Heider 1992 ble det vist at denne mutasjonen reduserer UGA-oversettelse med 99% (se Heider’s Figur 4A, kolonnen merket C3).
Baron fant at bindingen mellom SelB og mutant-RNA var betydelig svakere enn bindingen mellom SelB og u-mutert SECIS-RNA (se Baron’s Figur 1B). Dette indikerer at guanin på tredje posisjon i SECIS-løkken er involvert i bindingen mellom SECIS-RNA og SelB. Altså støttes hypotesen om at SelB binder til SECIS-løkken.
Klug, Huttenhofer, Kromayer, Famulok (1997): In vitro and in vivo characterization of novel mRNA motifs that bind special elongation factor SelB
Heider foreslo en modell for UGA-oversettelse hvor SelB binder til SECIS-løkken mens SECIS-stammen holder løkken (og dermed SelB + tRNASecACU) i riktig posisjon i forhold til ribosomet (se Heider 1992-b Figur 9). For å utfordre denne modellen forsøkte Klug å finne muterte SECIS-stammeløkker som både kan binde til SelB og som har riktig lengde på stammen (en lengde lignende stammen til SECIS-villtypen), men som samtidig IKKE fasiliterer UGA-oversettelse.
I motsetning til de tidligere forskningstekstene nevnt i denne artikkelen, hvor det ble laget en mindre antall SECIS-mutanter med bestemte mutasjoner, så laget Klug et stort antall SECIS-mutanter med tilfeldige mutasjoner. Mer nøyaktig så skriver han at det ble laget 5 × 1014 ulike mutanter, hvor den gjennomsnittlige SECIS-mutanten hadde tolv tilfeldig muterte baser. (Hele SECIS er 39 baser lang og det benyttes en tilfeldig mutasjonsrate på 30% per base, som betyr at den gjennomsnittlige mutanten vil ha 0.3 × 39 ≈ 12 muterte baser, men mange mutanter hadde flere eller færre enn tolv ettersom prosessen er tilfeldig).
SelB og SECIS-mutanter ble heldt over et filter med porer på 0.45 mikrometer. SelB er for stort til å gå gjennom porene, så SECIS-mutanter bundet til SelB ble værende i filteret, mens ubundede SECIS-mutanter gikk gjennom. For å bestemme sekvensene til de SelB-bindende SECIS-mutantene ble RNA fra filteret revers-transkriptert til DNA før det ble DNA-sekvensert.
Sekvenseringen identifiserte tretti ulike SECIS-mutanter som kunne binde SelB. Nitten av mutantene inneholdt en «invariant region» i den apikale delen av SECIS (fra base G16 til C30), hvor mutantenes baser er identiske med SECIS-villtypens baser (se Klug’s Figur 1B, SECIS-villtypen (wt fdhF) vises på topp mens de ulike mutantene refereres til som "clones").
De resterende elleve mutantene hadde større sekvens-variasjon i den apikale delen i forhold til SECIS-villtypen (se Klug's Figur 1C). Kun tolv av de tretti mutantene ble brukt i videre forsøk: en mutant med invariant region, samt alle elleve mutantene med større sekvens-variasjon.
Videre forsøk
Det ble undersøkt hvor sterkt de tolv utvalgte SECIS-mutantene binder til SelB. Hver individuelle mutant ble blandet med SECIS-villtypen og SelB, slik at konsentrasjonene til de tre var 10 nanomolar SelB, 40 nanomolar mutant, og 200 nanomolar villtype. Dette betyr at i hver prøve vil mutanter og villtyper "konkurrerer" om binding til en begrenset mengde SelB. (Jeg vet ikke hvorfor det ble brukt mer villtype enn mutant, synes det virker mer logisk å bruke en lik mengde av begge.) Prøvene ble heldt over 0.45 mikrometer filtere.
For å bestemme bindingsratioen til de to RNA i filteret, altså antall SelB-bundede mutanter per SelB-bundet villtype, så ble mutantene og villtypene først separert fra hverandre med gelelektroforese (dette var mulig fordi de to RNA var av ulike lengder). Før de to RNA ble blandet sammen med SelB var begge gjort radioaktive ved å feste en radioaktiv fosfat til 5'-enden av hvert individuelle RNA-molekyl. Den relative mengden av mutant og villtype i hver prøve kunne derfor måles ut ifra radioaktivitetet i de to gele-båndene som inneholdt de to RNA.
Bindingsratioen for hver av de tolv mutantene vises i Klug’s Tabell 1. Mutant #945, den som inneholdt en invariant region, hadde en bindingsratio på 2.0 (tyve SelB-bundede mutant-RNA per ti SelB-bundede villtype-RNA i filteret). De elleve andre mutantene hadde bindingsratioer mellom 0.1 til 3.0. Altså er det noen SECIS-mutanter som binder SelB bedre enn SECIS-villtypen.
I et siste forsøk ble fire SelB-bindende SECIS-mutanter, #945, #922, #934, og #979, undersøkt for evne til å fasilitere UGA-oversettelse (bindingsratioene var henholdsvis 2.0, 0.5, 0.8, og 0.7). Heider 1992-b viste at SECIS-stammens lengde var viktig for UGA-oversettelse, og Klug kommenterer at de fire utvalgte mutantene kan danne stammer med omtrent samme lengde som villtypen (se Klug’s Figur 4A). UGA-oversettelse ble målt via β-galaktosidase-aktivitet som i Zinoni 1990.
Beregnede Miller-verdier for de fire SECIS-mutantene samt villtypen er gitt i Klug’s Tabell 2. Vi ser at mutant #945 (den ene av de fire mutantene med invariant region) har en verdi på 970, mens villtypen (WT 60.2) har 1100. Altså kan mutant #945 fasilitere UGA-oversettelse ganske bra. I de andre tre mutantene #922.1, #934.1, og #979.1 er Miller-verdiene nær null, så UGA-oversettelse fungerer ikke med disse mutantene. Dette viser at binding av SelB til en SECIS-stammeløkke med riktig lengde på stammen ikke i seg selv er nok til å fasilitere oversetting av UGA.
En utvidet modell
Som nevnt i starten av denne seksjonen så presenterte Heider en mulig modell for UGA-oversettelse hvor SelB binder til SECIS-løkken mens SECIS-stammen holder løkken i riktig posisjon i forhold til ribosomet.
Ut i fra sitt resultat foreslår Klug en en utvidelse av denne modellen. Han tenker at SelB-proteinet kan eksistere i en aktiv og en inaktiv form, hvor den inaktive SelB kan binde til tRNASec og til SECIS-løkken, men kan ikke oversette UGA. Kun den aktive formen av SelB kan binde til ribosomet og tillate oversetting av UGA.
I den utvidede modellen er SECIS-stammeløkkens evne til å binde og holde SelB på korrekt avstand fra ribosomets A-sted ikke nok til å skape UGA-oversettelse. I tillegg må stammeløkken ha en struktur som induserer aktivering av en bundet SelB. En slik aktiveringsmekanisme kan tenkes å være nyttig, siden den kan forhindre at SelB-tRNASec-komplekset feilaktig binder til en UGA som er ment å være en stopp-kodon (i hvilket tilfelle det ikke vil være noen SECIS-stammeløkke som kan aktivere SelB).
Liu, Reches, Groisman, Engelberg-Kulka (1998): The nature of the minimal “selenocysteine insertion sequence” (SECIS) in Escherichia coli
SECIS-stammen i E. coli's fdhF-gen består av fjorten basepar (inkludert et mulig GU-par) fra A3-U42 nederst i stammen til C20-G27 øverst i stammen (basene er nummerert fra første base i UGA-kodonet, se Liu's Figur 1A hvor SECIS-villtypen er merket "a").
Baron 1993 viste at SelB kan binde til RNA som inneholder SECIS-stammeløkken. Deretter viste Kromayer 1996 at SelB's domene 4b kan binde til en mini-SECIS-stammeløkke, bestående av SECIS-løkken og en stamme med kun fem basepar, fra G15-C31 til C20-G27. (Kromayer testet ikke om mini-SECIS var funksjonell i UGA-oversetting.)
Resultatene til Zinoni 1990 indikerte at effektiv UGA-oversettelse er avhengig av alle fjorten baseparene i villtype SECIS-stammen. I lys av Kromayer's resultat ønsket Liu å undersøke på nytt om alle fjorten baseparene er nødvendige for UGA-oversettelse, eller om en SECIS-stamme med færre basepar kan fasilitere effektiv UGA-oversettelse. For å lage stammer med redusert lengde ble bestemte baser i den ene halvparten av SECIS mutert slik at de var identiske med motsatte baser i den andre halvparten av stammen. For eksempel, et CG-par kan muteres til G og G. Dette forhindrer bestemte basepar fra å dannes.
I SECIS-mutant 1b ble de fire baseparene fra C4-G41 til G8-U38 forhindret, dette førte til en ≈ 20% reduksjon av β-galaktosidase-aktivitet kontra SECIS-villtypen (se Liu's Figur 1). I mutant 1c ble de åtte baseparene fra C4-G41 til U13-A33 forhindret, med samme 20% reduksjon i β-galaktosidase-aktivitet. I mutant 1d var ni basepar brutt, hvor det niende baseparet var et G15-C31-par som i mutanten ble omgjort til C15 og C31. Dette resulterte i 90% reduksjon av β-galaktosidase-aktivitet. Det ble dermed vist at UGA-oversettelse kunne fungere ganske godt (men ikke optimalt) med mutant 1c sin mini-stamme bestående av kun fem basepar fra G15-C31 til C20-G27.
Angående mutant 1d, så ble det tenkt at reduksjonen i UGA-oversettelse enten kom av mangelen på baseparing av basene på posisjon 15 og 31, eller alternativt, kom av at det ikke var guanin på posisjon 15. Det ble derfor laget en mutant 1e hvor villtypens G15-C31-par ble "snudd" til et C15-G31-par. I denne mutanten kan basene på posisjon 15 og 31 basepare, samtidig som at det ikke er guanin på posisjon 15. I denne mutanten var β-galaktosidase-aktiviteten bare redusert 20% kontra SECIS-villtypen, samme reduksjon som i mutanter 1b og 1c (se Liu’s Figur 1). Altså trenger det ikke å være guanin på posisjon 15.
Med utgangspunkt i mutant 1c med mini-stammen på fem basepar ble det laget fire nye SECIS-mutanter for å teste hvordan lengden mellom UGA og mini-stammen påvirker UGA-oversettelse. En mutant hadde tre baser fjernet mellom UGA og mini-stammen, en annen hadde seks baser fjernet, en tredje hadde tre ekstra baser satt inn mellom UGA og mini-stammen, og en fjerde hadde seks ekstra baser satt inn (se Liu's Figur 2).
Det ble funnet at både innsetting og fjerning av tre baser førte til en ≈ 50% reduksjon i β-galaktosidase-aktivitet kontra SECIS-villtypen, mens innsetting eller fjerning av seks baser førte til en >90% reduksjon i β-galaktosidase-aktivitet. Det er altså tydelig at lengden mellom UGA og mini-stammen er av betydning for UGA-oversettelse, og det kan se ut som om villtype-lengden (elleve baser) er optimal.
Den utstikkende uracil
SECIS-stammen inneholder to uraciler (U17 og U18) posisjonert motsatt av èn enkelt adenin (A29). Dette betyr at en av de to basene ikke kan delta i et basepar og må istedet stikke ut til siden av stammen. Spørsmålet var da: er en utstikkende base på dette stedet viktig for UGA-oversettelse? Isåfall, må den utstikkende basen nødvendigvis være på posisjon 17 eller på posisjon 18, eller kan begge posisjonene benyttes? Og må den utstikkende basen nødvendigvis være en uracil, eller kan adenin, cytosin, eller guanin også fungere?
For å besvare første og andre sprøsmål ble det laget fire SECIS-mutanter. I mutant 3b var U18 endret til A18, slik at et eventuelt basepar mellom posisjoner 18 og 31 forhindres (men U17 og A31 kan fremdeles basepare). Denne mutanten vil altså ha en utstikkende adenin på posisjon 18. I mutant 3d var U17 endret til A17, slik at et eventuelt basepar mellom posisjoner 17 og 31 forhindres (men U18 og A31 kan fremdeles basepare). Denne mutanten vil ha en utstikkende adenin på posisjon 17. Begge mutantene hadde en >90% reduksjon i β-galaktosidase-aktivitet kontra villtypen (se Liu’s Figur 3).
I mutant 3c var det mulige U18-A31-paret i villtype-SECIS snudd til et A18-U31-par, slik at det vil være en utstikkende uracil på posisjon 17. Og i mutant 3e var det mulige U17-A31-paret i villtypen snudd til et A17-U31-par, slik at det vil være en utstikkende uracil på posisjon 18. Mutanten med utstikkende U17 hadde en like høy β-galaktosidase-aktivitet som SECIS-villtypen, mens mutanten med utstikkende U18 hadde en >90% reduksjon i β-galaktosidase-aktivitet, lignende mutanter 3b og 3d (se Liu’s Figur 3).
UGA-oversettelse kan altså fungere med en utstikkende uracil på posisjon 17, men ikke med en utstikkende adenin på posisjon 17, ei heller med en utstikkende uracil eller adenin på posisjon 18.
Det ble deretter laget ytterlige to mutanter, hvor U17 ble endret til henholdsvis C17 og G17, for å undersøke om en utstikkende cytosin eller guanin på posisjon 17 kan fungere. Disse mutantene viste samme >90% reduksjon i β-galaktosidase-aktivitet som i mutanten hvor U17 ble endret til A17, så den utstikkende basen på posisjon 17 må nødvendigvis være en uracil.
Resultater som ikke samsvarer
Liu's resultat om at åtte basepar i den basale delen av SECIS-stammen kan forhindres uten mer enn 20% reduksjon av UGA-oversettelse strider mot resultatet fra Zinoni 1990. Zinoni fant at Mut-39, hvor kun ett basalt basepar er forhindret, førte til en 55% reduksjon i UGA-oversettelse (sammenlignet med Mut-46/47/75).
Om dette tenker jeg at det må ha vært noe i Zinoni's forsøk som negativt påvirket UGA-oversettelse i Mut-39. Det kan muligens være at den fusjonerte mRNA kan danne en alternativ stammeløkke. Dersom en del av SECIS kan inngå i en alternativ stammeløkke, så kan en mRNA enten danne SECIS-stammeløkken eller den alternative stammeløkken, ikke begge to samtidig. Eksistensen av en alternativ stammeløkke burde derfor kunne redusere UGA-oversettelse. Men dette er bare spekulasjon.
Videre har vi Liu's resultat om at fjerning av tre baser mellom UGA og mini-SECIS-stammen fører til 50% reduksjon i UGA-oversettelse. Dette strider litt med Heider 1992-b, hvor det ble vist at fjerning av tre basepar i den basale delen av SECIS-stammen førte til en nærmest 100% reduksjon i UGA-oversettelse. Her aner jeg ikke hva som kan være grunnen til uoverstemmelsen.
Sandman, Tardiff, Neely, Noren (2003): Revised Escherichia coli selenocysteine insertion requirements determined by in vivo screening of combinatorial libraries of SECIS variants
For å forstå Sandman's forsøk er det nødvendig å vite litt om et virus kalt M13 samt en metode kalt lacZ alfa-komplementering.
M13-virus består av DNA innkapslet i et protein-skjell (kapsid) som er bygd opp av fem ulike proteiner kalt pIII, pVI, pVII, pVIII, og pIX. Dersom en M13 binder til overflaten av en E. coli-celle kan virusets DNA entre cellen. Kapsid-protein pIII er essensielt for denne entringsprosessen. Inne i E. coli-cellen vil virusets DNA replikeres, og virusets gener transkripteres til virale mRNA som oversettes til virale proteiner. Virale proteiner og DNA settes så sammen til nye virus som sendes ut av E. coli-cellen for å infisere andre celler. (Detaljene om M13-viruset er fra Smeal 2016. Smeal refererer til de virale proteinene som p3 istedet for pIII, p6 istedet for pVI, etc. Sandman bruker navnet pIII, så i denne seksjonen bruker også jeg det navnet.)
lacZ er et gen som oversettes til β-galaktosidase, proteinet som bryter ned β-galaktosider som laktose. Via metoder for genetisk manipulering er det mulig å dele opp dette genet i to mindre gener: lacZΔM15 og lacZα (lacZ-alfa). Hverken av disse genene kan oversettes til en funksjonell β-galaktosidase, men de to mindre proteinene som oversettes fra lacZΔM15 og lacZα kan binde seg sammen til en funksjonell β-galaktosidase. Dette kalles alfa-komplementering.
Metode
I Liu 1998 ble det vist at SECIS-løkken og den apikale delen av SECIS-stammen kan fisilitere rimelig effektiv UGA-oversettelse selv i fravær av åtte basepar i den basale delen av SECIS-stammen. Altså er det betydelig toleranse for sekvens-variasjon i den basale delen av stammen.
Sandman undersøkte hvor mye sekvens-variasjon som tillates i den apikale delen av stammen, gitt at den basale delen av stammen i stor grad er baseparet. For Sandman's undersøkelse ble det laget SECIS-mutanter med tilfeldige mutasjoner i den apikale delen av stammen, fra U13-A33 til G19-C28 (se Sandman's Figur 3) (metoden for å lage tilfeldige mutasjoner var den samme som i Klug 1997).
Hver mutant ble satt inn i en M13-DNA, i begynnelsen av genet for pIII-proteinet. Et M13-virus' evne til å produsere pIII-proteiner og infisere nye E. coli-celler er da avhengig av at viruset har en SECIS-mutant som tillater UGA-oversettelse. (På samme måte som at produksjon av fdhF-lacZ-fusjonsproteiner i Zinoni 1990 var avhengig av at fdhF-mutanten tillot UGA-oversettelse.)
De genmodifiserte M13 ble satt inn i E. coli-celler. Disse infiserte E. coli ble dyrket på en næringsgele sammen med ikke-infiserte E. coli. At bakteriene dyrkes i en næringsgele istedetfor i en flytende næringsløsning betyr at bakteriene ikke kan bevege seg rundt omkring, de forblir på stedet de var da geleen stivnet.
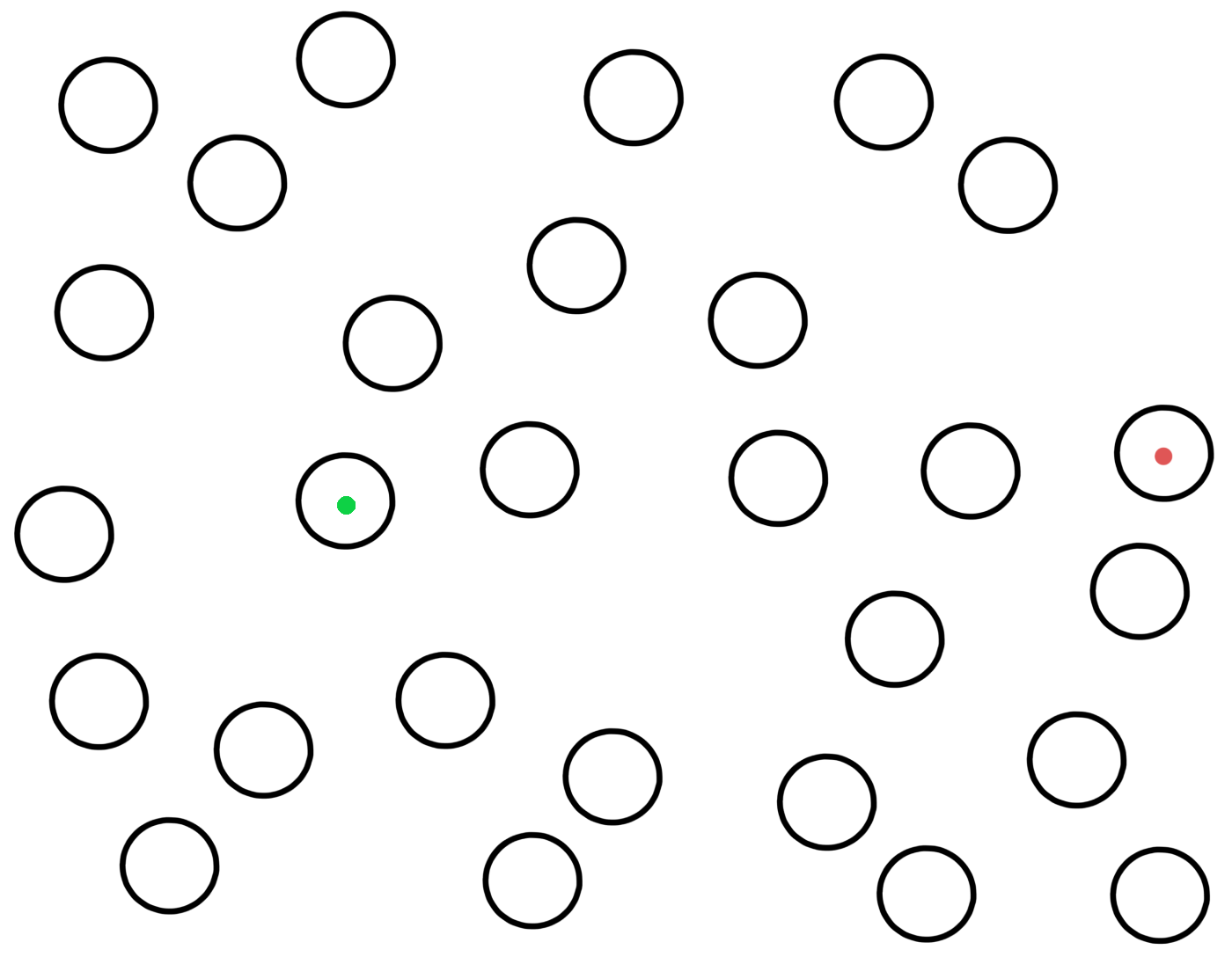
En M13 med SECIS-mutant som fasiliterer UGA-oversettelse vil produsere nye virus som slippes ut av den infiserte E. coli og infiserer andre E. coli i nærheten (virus er små nok til å diffusere gjennom næringsgeleen). Disse andre infiserte E. coli produserer så flere virus som infiserer flere E. coli i nærheten, og på denne måten vil infeksjonen spre seg som en voksende sirkel sentrert på den opprinnelige infiserte cellen.
Næringsgeleen som E. coli-cellene dyrkes på inneholder β-galaktosider kalt X-gal. β-galaktosidase kan bryte ned X-gal til galaktose og 5,5’-dibromo-4,4’-dikloro-indigo, hvor sistnevnte er et blåfarget molekyl.
E. coli-cellene i geleen har ikke et vanlig lacZ-gen, men har istedet et lacZΔM15-gen, mens DNA fra M13-viruset inneholdt et lacZα-gen. Dette betyr at infiserte E. coli kan produsere funksjonelle β-galaktosidaser via alfa-komplementering, og β-galaktosidasene kan bryte ned X-gal til et blåfarget molekyl, slik at infiserte E. coli blir blå. Ikke-infiserte E. coli forblir uten farge. Infiserte celler kan derfor ses som blå prikker i næringsgeleen. M13-DNA fra disse prikkene ble sekvensert for å identifisere de SECIS-mutantene som muliggjør UGA-oversettelse.
X-gal vs ONPG
I Sandman's forsøk testes β-galaktosidase-aktivitet med X-gal, som brytes ned til det blå molekylet dibromo-dikloro-indigo, mens i Zinoni 1990 ble β-galaktosidase-aktivitet testet med ONPG, som brytes ned til det gule molekylet o-nitrophenol. Det er mer enn fargen som skiller disse to: o-nitrophenol er vannløselig, mens dibromo-dikloro-indigo er ikke det.
Når Sandman benytter X-gal til å lokalisere infiserte E. coli-celler i næringsgeleen så vil dibromo-dikloro-indigo forbli på stedet det ble produsert, sånn at kun infiserte E. coli-celler blir blå. Slik jeg forstår det, dersom Sandman hadde benyttet ONPG istedetfor X-gal, så ville hele næringsgeleen blitt gul og det ville vært umulig å lokalisere infiserte celler. Dette fordi o-nitrophenol er vannløselig og følgelig kan diffusere gjennom geleen (som er vannbasert). Dette skjer ikke med dibromo-dikloro-indigo fordi dette molekylet ikke er vannløselig.
I Zinoni's forsøk var det derimot nødvendig å bruke ONPG. I hans forsøk ble bakterienes β-galaktosidase-aktivitet testet ved å dyrke bakteriene i flytende (vannbaserte) næringsløsninger og bruke et spektrofotometer til å måle absorbansen i løsningene. Ettersom o-nitrophenol er vannløselig vil disse molekylene spre seg jevnt gjennom næringsløsningene som bakteriene vokser i. Dette er en forutsetning for at spektrofotometeret skal kunne estimere konsentrasjonen av o-nitrophenol. Dersom Zinoni hadde benyttet X-gal istedetfor ONPG, så tror jeg dibromo-dikloro-indigo ville ha klumpet seg sammen, ettersom dette molekylet ikke er vannløselig. Dette ville ha gjort det umulig å estimere molekylets konsentrasjon med et spektrofotometer.
Resultat
Det ble identifisert nitten ulike SECIS med mutasjoner i den apikale delen av stammen (se Sandman's Figur 3). Graden av UGA-oversettelse i hver mutant ble testet på lignende vis som i Zinoni 1990 (se Sandman's Figur 5).
(En kommentar til Sandman's Figur 5: Røde barer representerer verdier fra bakterier dyrket i medium med selenium. Blå barer representerer kontroll-verdier fra bakterier dyrket uten selenium, hvor UGA ikke kan oversettes til selenocystein (UGA kan fremdeles oversettes til tryptophan, selv om dette er lite effektivt, så det kan dannes en liten mengde funksjonelle β-galaktosidaser selv når UGA ikke oversettes til selenocystein). SECIS-villtypen refereres til som "TGACwt". β-galaktosidase-aktiviteten fra bakterier med SECIS-villtypen, dyrket med selenium, brukes som referanse-verdi. Den røde baren til villtypen viser derfor en verdi på 1 (= 100% β-galaktosidase-aktivitet). β-galaktosidase-aktivitet i SECIS-mutantene er oppgitt relativt til denne referanseverdien, slik at for eksempel en verdi på 1.4 betyr at β-galaktosidase-aktiviteten er 40% høyere enn i villtypen.)
Liu 1998 viste at SECIS' utstikkende U17 er viktig for UGA-oversettelse, og at denne basen ikke kan erstattes verken av en utstikkende U18 eller av en utstikkende A17/C17/G17. I Sandman's SECIS-mutanter SV15 og SV17 har villtypens U17 blitt endret til en G17, samtidig som at G16 har blitt endret til U16. Altså kan en utstikkende U16 erstatte en utstikkende U17, dog med redusert oversettelse av UGA (omtrent 50% reduksjon i variant SV15 og 85% reduksjon i variant SV17, relativt til SECIS-villtypen).
Videre viser Sandman's resultat at UGA kan oversettes effektivt selv om ikke alle de apikale basene i SECIS-stammen er baseparet. Atten av mutantene har ett forhindret basepar blant de fem baseparene nærmest løkken, og mange av disse mutantene fasiliterer høy UGA-oversettelse. I en mutant, SV13, forhindres tre av de fem baseparene nærmest løkken. Denne mutanten har en lavere grad av UGA-oversettelse enn de fleste andre mutantene.
Sammen med resultatet til Liu 1998 så viser Sandman's resultat at baseparing i den apikale delen av SECIS-stammen er viktigere enn baseparing i den basale delen av stammen, men den apikale delen trenger ikke nødvendigvis å være fullstendig baseparet for å oppnå høy UGA-oversettelse.
Oppsummering
Det ville sikkert vært mulig å skrive mye mer om SECIS, men jeg føler at denne artikkelen nå beskriver SECIS i E. coli ganske bra og stopper derfor her. Til avslutning, en liten oppsummering av det som har blitt presentert:
Et UGA-kodon kan fungere enten som et stopp-kodon eller som et selenocystein-kodon. For å fungere som et selenocystein-kodon er det nødvendig at UGA etterfølges av en SECIS (selenocysteine insertion sequence). I E. coli's fdhF-gen består SECIS av 39 baser like nedstrøms for UGA. SECIS kan danne en stammeløkke hvor løkkens seks baser samt en uracil som stikker ut fra stammen ser ut til å utgjøre kontakt-punktene ansvarlig for å binde til og aktivere SelB. Den aktiverte SelB sørger så for at tRNASec binder til UGA. SECIS-stammen må ha en viss lengde, ikke for kort og ikke for lang, for å posisjonere SelB korrekt i forhold til UGA. Stammen trenger ikke å være fullstendig baseparet, spesielt ikke den basale delen av stammen.
En detaljert forklaring av Miller's metode for måling av β-galaktosidase-aktivitet
Denne seksjonen er et tillegg til den tidligere seksjonen om Zinoni 1990.
"Miller-metoden" for å teste β-galaktosidase-aktivitet i en flytende cellekultur ble opprinnelig beskrevet i boken “Experiments in Molecular Genetics” (Jeffrey Miller, 1972). I korte trekk så kan metoden oppsummeres slik: bland inn ONPG (o-nitrophenyl-galactoside) in en flytende E. coli-kultur, vent til en merkbar gul farge oppstår i kulturen, og mål kulturens "optical density" ved 420 nanometers bølgelengde (altså: mål OD420). Miller brukte denne "aktivitet-formelen" for å beregne β-galaktosidase-aktiviteten:
Aktivitets-enheter (normalt kalt Miller-enheter) =
1000 × (OD420 – 1.75 × OD550) / (tid × volum × OD600)
For å forstå denne formelen er det nødvendig å forstå Miller's metode i mer detalj. Først blir E. coli-celler dyrket inntil de vokser raskt (inntil de er i den eksponensielle delen av vekstkurven). Glasset med kulturen plasseres så i is-vann for å avkjøle bakteriene og inhibere videre vekst. 1 mL av den avkjølte kulturen brukes for å bestemme celle-konsentrasjonen ved å måle OD600 i et spektrofotometer. (Cellekonsentrasjonen er proporsjonal med verdien av OD600.) Den målte OD600 settes inn i aktivitet-formelen.
Et annet volum fra den avkjølte kulturen blandes sammen med en løsning kalt "Z-buffer". Dersom det forventes at bakteriene vil ha en høy β-galaktosidase-aktivitet, bland 0.1 mL av den avkjølte kulturen med 0.9 mL Z-buffer. Dersom det forventes at bakteriene vil ha en lav β-galaktosidase-aktivitet, bland 0.5 mL av den avkjølte kulturen med 0.5 mL Z-buffer. Blandingen av bakterier og Z-buffer vil refereres til som "reaksjonsmiksen". Volumet av avkjølt kultur som ble brukt i reaksjonsmiksen settes inn i aktivitet-formelen.
(Miller forklarer ikke funksjonen til Z-buffer, but ifølge denne protokollen så er Z-buffer ment til å stabilisere β-galaktosidasene (forhindre at de inaktiveres over tid). Stabiliseringen kommer av en komponent i Z-buffer, β-mercapto-enthanol, som fungerer som en anti-oksidant og beskytter β-galaktosidasene mot reaksjon med oksygen. Jeg antar at en slik reaksjon vil redusere β-galaktosidasene's aktivitet.)
En dråpe toluen iblandes reaksjonsmiksen. Miller skriver at “The toluene partially disrupts the cell membrane, allowing small molecules such as ONPG to diffuse into the cells.” (Så jeg antar at toluen forårsaker dannelsen av små hull i cellemembranene.)
Deretter økes temperaturen i reaksjonsmiksen til 28°C, ONPG iblandes, og reaksjonens start-tid noteres. β-galaktosidasene i E. coli-cellene vil begynne å bryte ned ONPG til galaktose og o-nitrophenol, hvor sistnevnte er et gulfarget molekyl. Så snart en merkbar gul farge har oppstått i reaksjonsmiksen (noe som kan ta fra minutter til timer), så iblandes natrium-karbonat i reaksjonsmiksen. Natriumkarbonat endrer pH i reaksjonsmiksen fra pH 7 til pH 11, noe som forårsaker inaktivering av β-galaktosidasene og forhindrer videre nedbrytning av ONPG, slik den gule fargen ikke fortsetter å øke i intensitet. Reaksjonens stopp-tid noteres. Reaksjonens start-tid og stopp-tid brukes for å beregne reaksjonstiden (i minutter), som settes inn i aktivitet-formelen.
(Hvorfor utføres reaksjonen ved 28°C istedet for en annen temperatur, for eksempel 37°C? Sistnevnte temperatur ville økt reaksjonshastigheten. Jeg er ikke sikker, men jeg gjetter at β-galaktosidaser er betydelig mer stabile ved 28°C enn ved 37°C, og at Miller velger 28°C av denne grunn. Merk også at selv om temperaturen økes til 28°C så kan ikke E. coli fortsette å vokse, fordi cellemembranene deres har hull etter toluen-behandlingen. Cellekonsentrasjonen i reaksjonsmiksen vil derfor forbli konstant gjennom reaksjonen.)
Ved pH 11 vil o-nitrophenol absorbere lys mest effektivt ved 420 nanometers bølgelengde (absorbans-spekteret til o-nitrophenol vises i Figur 2 i denne PDF-en). Konsentrasjonen av o-nitrophenol i reaksjonsmiksen er proporsjonal med hvor mye lys reaksjonsmiksen absorberer ved 420 nm. Derfor måles reaksjonsmiksens OD420 (optical density ved 420 nm) i et spektrofotometer.
Lys-absorbsjon via o-nitrophenol er dog ikke den eneste faktoren som påvirker reaksjonsmiksens OD420. Lys-spredning via materiale fra bakterie-celler (“cell debris” som Miller kaller det) bidrar også til OD420-verdien. Vi kan derfor tenke på OD420 som en sum av ODabsorbert420 and ODspredt420, hvor det er ODabsorbert420 som vi ønsker å finne.
ODtotal420 = ODabsorbert420 + ODspredt420
Når OD måles ved en bølgelengde på 550 nm vil det være null absorbsjon fra o-nitrophenol (som kan ses på absorbans-spekteret linket ovenfor). Kun lysspredning fra cellemateriale vil bidra til verdien av OD550.
ODabsorbert550 = 0
ODtotal550 = ODspredt550
Graden av lysspredning fra cellemateriale er ikke den samme når en måler OD ved 420 og 550 nm bølgelengder. Ifølge Miller,
ODspredt420 = 1.75 × ODspredt550
Ved å kombinere ligningene ovenfor får vi:
ODabsorbert420 = ODtotal420 – 1.75 × ODtotal550
Derfor blir OD420 – 1.75 × OD550 satt inn i aktivitet-formelen.
Til sist blir brøken (OD420 – 1.75 × OD550) / (tid × volum × OD600) ganget med 1000. Ved å gjøre dette vil celler med svært lav β-galactosidase-aktivitet gi en verdi på omtrent 1 Miller-enhet. (Dersom vi ikke ganger med 1000 så vil celler med svært lav β-galaktosidase-aktivitet gi en verdi på omtrent 0.001 Miller-enheter. Jeg antar Miller foretrekker heltall over desimaler og at dette er grunnen til at det ganges med 1000 i aktivitet-formelen.)
Meningen med uttrykket tid × volum × OD600: hva aktivitet-enheter representerer
Vi kan nå se på hva aktivitet-formelen virkelig betyr. Uttrykket OD420 – 1.75 × OD550 er proporsjonalt til konsentrasjonen av o-nitrophenol i reaksjonsmiksen på det tidspunktet reaksjonen ble stoppet. Uttrykket (OD420 – 1.75 × OD550) / tid er proporsjonalt med økningen i o-nitrophenol-konsentrasjon per minutt mens reaksjonen pågår. Denne økningen i konsentrasjon er selv proporsjonel med β-galaktosidase-aktiviteten i reaksjonsmiksen.
OD600-verdien er proporsjonal med celle-konsentrasjonen (antall celler per volum) i bakterie-kulturen, så uttrykket volum × OD600 er proporsjonalt med antallet E. coli-celler som er til stede i reaksjonsmiksen. Uttrykket (OD420 – 1.75 × OD550) / (tid × volum × OD600) er derfor proporsjonalt med β-galaktosidase-aktiviteten per E. coli-celle i reaksjonsmiksen. Det er dette aktivitet-formelen beregner.
Eksempel-beregning av β-galaktosidase-aktivitet
Si at vi lar en E. coli-kultur vokse inntil den måler en OD600 på 0.562. Vi forventer en høy grad av β-galaktosidase-aktivitet og bruker derfor et kultur-volum på 0.1 mL i reaksjonsmiksen. Vi starter reaksjonen og sjekker fargen på reaksjonsmiksen hvert 15. minutt. Etter 75 minutter har en merkbar gul farge oppstått, og reaksjonen stoppes. Vi måler en OD420 på 0.833 og en OD550 på 0.102. β-galaktosidase-aktiviteten kan da beregnes til
1000 × (0.833 – 1.75 × 0.102) / (75 × 0.1 × 0.562) = 155 Miller-enheter
Alternativ aktivitet-formel uten OD550-måling
Miller bemerker at etter reaksjonen er stoppet med å iblande natrium-karbonat, så er det mulig å sentrifugere reaksjonsmiksen for å separere cellematerialet fra væsken. Cellematerialet vil samle seg i en liten "pellet" i bunnen av glasset som inneholder reaksjonsmiksen. Ved å legge til dette sentrifugering-steget til prosedyren så burde ODspredt420 være lik 0, slik at uttrykket – 1.75 × OD550 ikke lenger er nødvendig. Aktivitet-formelen blir da:
Miller-enheter = 1000 × OD420 / (tid × volum × OD600)
(Jeg har en følelse av at denne alternative versjonen av metoden og formelen bør beregne β-galaktosidase-aktiviteten mer nøyaktig, og å sentrifugere reaksjonsmiksen tar ikke mye tid, så jeg vet ikke hvorfor dette ikke er standard-formelen.)
Mengden ONPG som iblandes reaksjonsmiksen er viktig
Når vi bruker Miller-metoden for å bestemme β-galaktosidase-aktivitet så må aktiviteten være proporsjonal med antallet β-galaktosidaser per E. coli-celle. For at dette skal være sant er det nødvendig at alle β-galaktosidasene kontinuerlig bryter ned ONPG. Altså, β-galaktosidasene må være 100% mettet med ONPG. Derfor er det nødvendig at konsentrasjonen av ONPG som iblandes reaksjonsmiksen er mye høyere enn konsentrasjonen av β-galaktosidaser i reaksjonsmiksen.
Dersom kun en liten mengde ONPG iblandes, så vil kun en avdel av β-galaktosidasene være mettet. Graden av metning vil kanskje være 1% (altså: ved et hvert øyeblikk vil 1% av alle β-galaktosidasene holde på med nedbrytning av ONPG). Eller metningen vil kanskje være 90%, eller noe annet. Uten å vite metningsgraden kan vi ikke bruke β-galaktosidase-aktivitet som et indirekte mål for antallet β-galaktosidaser.
Miller iblander 0.2 mL ONPG-løsning (4 mg ONPG per mL vann) i 1 mL reaksjonsmiks. Med måten han utfører forsøket på er dette nok til å få 100% metning av β-galaktosidasene.
(Dersom en bruker Miller-metoden for å måle β-galaktosidase-aktivitet i et annet forsøk så vil det være enkelt å undersøke om 4 mL ONPG per mL vann gir en ONPG-løsning med høy nok konsentrasjon for å mette β-galaktosidasene fullstendig. Du kan lage to reaksjonsmikser, iblande 0.2 mL av en 4 mg ONPG-løsning i den ene reaksjonsmiksen, og iblande 0.2 mL av en 6 mg ONPG-løsning i den andre reaksjonsmiksen. Dersom løsningen med 4 mg ONPG er nok til å gi 100% metning av β-galaktosidasene, så vil også løsningen med 6 mg ONPG gi 100% metning, og de to reaksjonsmiksene vil ha samme verdi av OD420. Dersom de to reaksjonsmiksene gir ulike målinger av OD420 så viser dette at 4 mg-løsningen, og kanskje også 6 mg-løsningen, ikke fullstendig metter β-galaktosidasene. Du må da lage mer konsentrerte løsninger av ONPG og se om disse gir samme verdi av OD420.)
En forklaring av forsøksmetoden benyttet i Zinoni, Heider, Bock (1990): Features of the formate dehydrogenase mRNA necessary for decoding of the UGA codon as selenocysteine
Denne seksjonen er et tillegg til den tidligere seksjonen om Zinoni 1990. Det siteres fra Zinoni's "Materials and methods".
For generation of 3’ deletions of the fdhF gene, plasmid pFM22 was constructed by deletion of a 1.86 kilobase EcoRV/Xba I fragment from plasmid pFM30, thereby introducing a Bgl II site at codon 268 of fdhF.
Plasmid pFM30 inneholder en kopi av fdhF-genet (ifølge Zinoni’s referanse 9). I fdhF utgjør kodon nummer 269 og 270 en EcoRV restriction site (for å finne ut dette sjekket jeg gensekvensen til fdhF i BioCyc’s database). 1860 baser unna denne EcoRV-siten, nedstrøms langs fdhF-genet's nontemplat-DNA, finnes en Xba I restriction site.
5'...TTGAAGATATCACCGG...(1850 baser)...XXXXXTCTAGAXXXXX...3'
3'...AACTTCTATAGTGGCC...(1850 baser)...XXXXXAGATCTXXXXX...5'
(Jeg kjenner ikke sekvensen rundt Xba I-siten, derfor er basene i denne delen av sekvensen symbolisert med "X".)
Ved bruk av EcoRV og Xba I restriksjonsnukleaser kuttet Zinoni plasmid pFM30 på de to nevnte restriction sites slik at det ble dannet to fragmenter: ett fragment (med 1860 baser) inneholdt 3’-delen av fdhF-genet, mens et annet fragment inneholdt 5’-delen av fdhF-genet sammen med fdhF-promoteren.
3'...AACTTCTA TAGTGGCC...(1850 baser)...XXXXXAGATC TXXXXX...5'
Fragmentet med 5’-delen av fdhF isoleres med gelelektroforese. Den fragment-enden som ble kuttet med EcoRV er en blunt end, mens enden som ble kuttet med Xba I er en 5’ overhanging end. Sistnevnte "fylles inn" og omgjøres til en blunt end ved bruk av DNA polymerase I (Klenow), slik at fragmentet har to blunt ends.
3'...AACTTCTA GATCTXXXXX...5'
Fragment-endene ligeres sammen slik at det dannes sirkulært DNA. På ligeringsstedet dannes det en Bgl II restriction site, denne går fra tredje base i fdhF’s kodon nummer 268 til andre base i kodon nummer 270.
5'...TTGAAGATCTAGAXXXXX...3'
3'...AACTTCTAGATCTXXXXX...5'
Resultatet er at det er dannet et nytt plasmid pFM22, omtrent 1860 baser kortere enn pFM30, hvor kodon 268 etterfølges av en Bgl II restriction site istedet for en EcoRV restriction site.
(For å være ærlig, selv om jeg vet hva som blir gjort her så forstår jeg ikke hvorfor det blir gjort, hvorfor det er fordelaktig å lage en plasmid pFM22 istedet for å bare utføre neste del av forsøket med plasmid pFM30.)
Plasmid pFM22 was linearized with Bgl II and then treated with exonuclease BAL-31 at a degradation rate of 50 base pairs per minute.
Ved bruk av Bgl II restriksjonsnuklease ble plasmid pFM22 kuttet i Bgl II restriction site, slik at plasmidet gjøres lineært. fdhF’s kodon 268 ligger ved den ene plasmid-enden og utgjør nå fdhF’s nontemplat 3’-ende. Eksonukleasen BAL-31 anvendes for å kutte bort baser fra plasmid-endene, slik at fdhF's nontemplat kuttes fra 3’-enden i retning mot 5’-enden.
Etter en bestemt tid inaktiveres BAL-31 ved å øke temperaturen, slik at kuttingen stopper nær fdhF’s UGA-kodon. Ved å bruke flere prøver av plasmidet og BAL-31 og variere tiden før inaktivering av BAL-31 kan det dannes forskjellige 3’ deletion-mutanter av fdhF's nontemplat.
fdhF’s UGA-kodon er kodon nummer 140, mens Bgl II restriction site ligger 3’ for kodon 268. Altså er det 128 kodoner, eller 384 baser, som skiller de to. Zinoni's mutant Mut-384 ble derfor produsert uten å bruke BAL-31 til å kutte plasmid-endene.
After «fill-in» of 5’ protruding ends with DNA polymerase I (Klenow), ligation with BamHI linkers (dCGCGGATCCGCG), and restriction with EcoRI and BamHI, the fragments were separated on a 1.5% agarose gel and recloned into EcoRI/BamHI-digested plasmid pACYC184. Inserts were sized by restriction analysis and their exact end points were determined by DNA sequencing.
Å kutte endene av plasmidet med BAL-31 kan danne både blunt ends og overhanging ends. 5’ overhanging ends fylles inn med DNA polymerase I (Klenow) slik at alle endene blir blunt ends. Begge ender av plasmidet ligeres til syntetiske «linker-DNA» som hver inneholder en BamHI restriction site. («d» foran basesekvensen til linkeren (dCGCGGATCCGCG) betyr «deoksy» og brukes for å spesifisere at linkeren er en DNA-sekvens, ikke en RNA-sekvens.)
For eksempel, en av 3' deletion-mutantene produsert av BAL-31 er Mut-7, hvor UGA-kodonet etterfølges av syv baser. Her blir Mut-7 ligert til en linker:
5'...TGACACGGCC CGCGGATCCGCG 3'
3'...ACTGTGCCGG GCGCCTAGGCGC 5'
Etter ligering:
5'...TGACACGGCCCGCGGATCCGCG 3'
3'...ACTGTGCCGGGCGCCTAGGCGC 5'
(Ettersom disse sekvensene er DNA, og DNA inneholder basen T istedet for U, så vises UGA-kodonet her som TGA.)
Etter ligering blir plasmiden kuttet med EcoRI og BamHI restriksjonsnukleaser. BamHI kutter i BamHI-siten i linkeren ved fdhF-genets forkortede 3’-ende, mens EcoRI kutter i en EcoRI restriction site som befinner seg et sted 5’ for fdhF-promoteren (jeg er ikke sikker på nøyaktig hvor langt unna promoteren denne restriction site er). Det resulterende plasmid-fragmentet med fdhF-promoteren og fdhF's 5'-ende isoleres med gelelektroforese.
Kuttene fra BamHI og EcoRI har gitt fragmentet to ulike overhanging ends. En pACYC184-plasmid kuttes av BamHI og EcoRI slik at dette plasmidet får samme overhanging ends som fdhF-fragmentet. Plasmidet blandes sammen med fragmentet slik at deres ender baseparer sammen og kan ligeres.
For å verifisere at fdhF-fragmentet ble satt inn korrekt i plasmid pACYC184 benyttes restriksjonsanalyse samt DNA-sekvensering av sekvensene rundt de to ligeringsstedene.
These fdhF fragments were used to generate lacZ fusions with one of plasmids pFM1400, pFM1401, and pFM1402. They carry the polylinker of plasmid pUC8 in front of a truncated lacZ gene in all three reading frames and differ in only 1 or 2 bases, thus minimizing effects arising from different lacZ linker joints.
Plasmidene pFM1400/1401/1402 inneholder et lacZ-gen hvor de første syv kodonene har blitt fjernet (ifølge Zinoni’s referanse 15). (Det er mulig å fjerne de første syv kodoner fra lacZ uten at det resulterende β-galaktosidase-proteinet mister sin funksjon.) På 5’-siden av lacZ finnes en polylinker, altså en basesekvens som inneholder mange restriction sites klynget sammen. Her er sekvensen til polylinkeren i plasmid pFM1401 foran lacZ-genet (som starter ved et kodon som korresponderer til åttende kodon i lacZ-villtypen):
/ \ / \ / \ lacZ (nontemplat)
5'...GAATTCCCGGGGATCCGTCGACCTGCAGCCAAGCTTGCTCTGGCCGTCGTTTTA...3'
3'...CTTAAGGGCCCCTAGGCAGCTGGACGTCGGTTCGAACGAGACCGGCAGCAAAAT...5'
\ / \ / \ /
SmaI AccI HindIII
SalI
Blant polylinkerens restriction sites er en EcoRI site og en BamHI site som brukes til å sette inn et fdhF-fragment foran lacZ, slik at det dannes et fusjonsgen. Hvert plasmid med fusjonsgen settes inn i E. coli-celler via elektroporering.
fdhF-fragmentene som settes inn i plasmidene mangler transkripsjonsterminator, ettersom denne har blitt fjernet sammen med fdhF’s 3’-ende. Videre så har ikke plasmidenes lacZ-gen noen lac-promoter. Dette betyr at i fusjonsgenet kan ikke fdhF-delen og lacZ-delen transkripteres individuelt, de kan kun transkripteres sammen som én mRNA.
fdhF har en start-kodon, mens lacZ har ikke dette. Oversetting må derfor starte ved fdhF. Oversettingen av fusjonsgenet kan stanse på to punkter: Dersom fdhF’s UGA-kodon ikke oversettes til selenocystein (på grunn av manglende SECIS-struktur) vil dette kodonet fungere som et stopp-kodon, og det resulterende proteinet inneholder kun en del av fdhF-proteinet. Alternativt, dersom UGA oversettes til selenocystein vil oversettingen stoppe ved lacZ’s stopp-kodon. (fdhF's stopp-kodon har blitt fjernet, så oversetting stopper ikke ved enden av fdhF.)
Det viktige poenget er at lacZ i plasmidene ikke kan oversettes individuelt, de kan kun oversettes som en del av fdhF-lacZ-fusjonsproteiner. Plasmider med fusjonsgener settes inn i E. coli-celler hvor det eksisterende lacZ-genet er defekt. Fusjonsproteinene vil derfor være ansvarlige for all β-galaktosidase-aktivitet i bakteriene, en nødvendighet for at β-galaktosidase-aktivitet skal være proporsjonal med konsentrasjonen av fusjonsproteiner. (Som nevnt tidligere så er dette nødvendig for at Miller-verdiene skal være proporsjonale med effektiviteten av UGA-oversettelse).
Nå om hvorfor fdhF-fragmenter settes inn i tre forskjellige plasmider pFM1400, pFM1401, og pFM1402. Alle basesekvenser kan leses som tre ulike basetriplett-sekvenser, kalt leserammer (reading frames). Her er et eksempel fra starten av lacZ-genet (nontemplat-DNA):
...GAA ACA GCT ATG ACC ATG ATT ACG GAT TCA CTG GCC GTC GTT TTA C...
...G AAA CAG CTA TGA CCA TGA TTA CGG ATT CAC TGG CCG TCG TTT TAC...
...GA AAC AGC TAT GAC CAT GAT TAC GGA TTC ACT GGC CGT CGT TTT AC...
I oversetting av en gensekvens til et protein benyttes kun den leserammen som inneholder start-kodonet:
1st 2nd 3rd 4th 5th 6th 7th 8th 9th 10t 11t 12t...
...GAA ACA GCT ATG ACC ATG ATT ACG GAT TCA CTG GCC GTC GTT TTA C...
Met Thr Met Ile Thr Asp Ser Leu Ala Val Val Leu ...
(korresponderende aminosyrer)
Når to gener settes sammen til et fusjonsgen er det nødvendig at genenes kodoner er i samme leseramme, fordi begge genene oversettes fra samme start-kodon.
fdhF og lacZ forbindes av en polylinker-sekvens, og lengden på denne er avgjørende for om de to genene plasseres i samme leseramme eller ikke. Fordi det brukes fdhF-mutanter med ulike 3’-deletions så brukes også tre polylinkere som varierer i lengde med en eller to baser (polylinker-sekvensene er gitt i Zinoni’s referanse 15). Hver av de tre polylinkerne finnes i sitt eget plasmid (en i pFM1400, en i pFM1401, en i pFM1402). Her er en sammenligning av de tre polylinker-sekvensene (den variable delen av polylinkerne er markert med fet tekst, lacZ-genets åttende kodon CTG starter rett nedstrøms av den variable delen):
5'...GAATTCCCGGGGATCCGTCGACCTGCAGCCAAGCTTGCGATCTGGCCGTCGTTTTA...3'
3'...CTTAAGGGCCCCTAGGCAGCTGGACGTCGGTTCGAACGCTAGACCGGCAGCAAAAT...5'
Polylinker i pFM1401:
5'...GAATTCCCGGGGATCCGTCGACCTGCAGCCAAGCTTGCTCTGGCCGTCGTTTTA...3'
3'...CTTAAGGGCCCCTAGGCAGCTGGACGTCGGTTCGAACGAGACCGGCAGCAAAAT...5'
Polylinker i pFM1402:
5'...GAATTCCCGGGGATCCGTCGACCTGCAGCCAAGCTTCGATCTGGCCGTCGTTTTA...3'
3'...CTTAAGGGCCCCTAGGCAGCTGGACGTCGGTTCGAAGCTAGACCGGCAGCAAAAT...5'
For å ta et praktisk eksempel på hvorfor det må brukes tre polylinkere: dersom fdhF-mutant Mut-7 settes inn i polylinkeren i plasmid pFM1401 vil lacZ’s kodoner oversettes til de korrekte aminosyrene (sammenlign med lacZ-kodonene og de korresponderende aminosyrene som ble vist tidligere):
fdhF lacZ
5'...TGACACGGCCCGCG GATCCGTCGACCTGCAGCCAAGCTTGCTCTGGCCGTCGTTTTA...3'
3'...ACTGTGCCGGGCGCCTAG GCAGCTGGACGTCGGTTCGAACGAGACCGGCAGCAAAAT...5'
Mut-7 ligert til pFM1401:
fdhF lacZ
5'...TGACACGGCCCGCGGATCCGTCGACCTGCAGCCAAGCTTGCTCTGGCCGTCGTTTTA...3'
3'...ACTGTGCCGGGCGCCTAGGCAGCTGGACGTCGGTTCGAACGAGACCGGCAGCAAAAT...5'
lacZ oversettes til korrekte aminosyrer: lacZ-kodoner
... ... ... ... ... ... ... ... ... ... ... ... ... ... ... 8th 9th 10t 11t 12t ...
5'... TGA CAC GGC CCG CGG ATC CGT CGA CCT GCA GCC AAG CTT GCT CTG GCC GTC GTT TTA ...3'
... Sec His Gly Pro Arg Ile Arg Arg Pro Ala Ala Lys Leu Ala Leu Ala Val Val Leu ...
Mens dersom Mut-7 settes inn i polylinkeren i plasmid pFM1400 eller pFM1402 vil lacZ oversettes til feil aminosyrer (markert med rød tekst):
5'... TGA CAC GGC CCG CGG ATC CGT CGA CCT GCA GCC AAG CTT GCG ATC TGG CCG TCG TTT ...3'
... Sec His Gly Pro Arg Ile Arg Arg Pro Ala Ala Lys Leu Ala Ile Trp Pro Ser Phe ...
Mut-7 satt inn i pFM1402:
5'... TGA CAC GGC CCG CGG ATC CGT CGA CCT GCA GCC AAG CTT CGA TCT GGC CGT CGT TTT ...3'
... Sec His Gly Pro Arg Ile Arg Arg Pro Ala Ala Lys Leu Arg Ser Gly Arg Arg Phe ...
Mut-18, derimot, må settes inn i plasmid pFM1402 for at lacZ skal kunne oversettes til riktig aminosyrer, mens Mut-47 må settes inn i plasmid pFM1400. De andre mutantene må også settes inn i en bestemt plasmid for at lacZ skal oversettes riktig.
(Angående hvorfor Zinoni setter fdhF-fragmenter inn i plasmid pACYC184 før de kuttes ut og settes inn i plasmider pFM1400/1401/1402, så er dette også noe jeg ikke forstår og ikke kan forklare.)
En forklaring av forsøksmetoden benyttet i Heider, Baron, Bock (1992-b): Coding from a distance: dissection of the mRNA determinants required for the incorporation of selenocysteine into protein
Denne seksjonen er et tillegg til den tidligere seksjonen om Heider 1992-b. Det siteres fra Heider's "Materials and methods".
Mutated DNAs [containing the selenocysteine codon and its accompanying selenocysteine insertion sequence] were generated in vitro by annealing either two or four oligonucleotides, filling in single-stranded regions and inserting the products into plasmid pUC9 or pT3T7lac.
In the case of the mutagenesis of the loop of the assumed stem-loop-structure, six separate fill-in reactions were performed. In each of the reactions one of six different oligonucleotides, each degenerate at one position of the loop, was used. It was annealed with three oligonucleotides providing the rest of the [DNA sequence] such that the site of degeneracy was situated in a single-stranded region.
For å produsere en SECIS med mutert løkkesekvens starter Heider med å bruke oligonukleotid-syntese til å bygge fire korte ssDNA (single-stranded DNA), hvor en av de fire inneholder ønsket mutasjon. Hver ssDNA har en basesekvens som gjør det mulig å basepare til en eller to av de andre ssDNA, slik at de sammen kan danne en DNA som har alternerende single-stranded og double-stranded regioner.
Nedenfor vises et eksempel på en slik DNA, hvor løkken er markert med fet tekst. SECIS-villtypen har løkkesekvens AGGTCT, så denne eksempel-DNA er en mutant hvor løkkens første base er endret fra A til G.
3'-TTCGAAAACTGTGCCGGGTAGCCAAp TGGTTAGCCAGCCAAAAACCTAGG-5'
The two oligonucleotides lying within the annealed product had been phosphorylated at their 5’-ends. The annealed product was treated simultaneously with T4 DNA polymerase and T4 DNA ligase to allow formation of complete double-stranded DNA.
De tre single-stranded regioner fylles inn med T4 DNA polymerase, og ligeres med T4 ligase slik at det dannes en komplett dsDNA (double-stranded DNA). (T4 DNA polymerase stammer fra T4-viruset og er velegnet til å fylle inn gaps.)
Nedenfor vises DNA etter fyll-inn og ligering, hvor TGA-kodonet ( = UGA i mRNA) er understreket og selenocysteine insertion sequence (SECIS) er markert med fet tekst.
3'-TTCGAAAACTGTGCCGGGTAGCCAACGCCCAGACGTGGTTAGCCAGCCAAAAACCTAGG-5'
For at ligering skal fungere er det nødvendig at 5’-endene som skal ligeres er bundet til fosfat. DNA som er bygd med oligonukleotid-syntese mangler fosfat på 5’-endene, så de to korte DNA hvis 5’-ender skal ligeres må derfor fosforyleres. Fosforylering utføres ved å blande DNA med ATP (adenosin trifosfat) og polynukleotid kinase, et enzym som kan overføre fosfat fra ATP til 5’-enden av DNA. I DNA-sekvensen ovenfor med alternerende single-stranded og double-stranded regioner er fosforylerte 5'-ender markert med "p".
In the case of mutagenesis of [the stem region of the stem-loop-structure], oligonucleotides were annealed pairwise and filled in using Klenow [DNA polymerase].
Mutasjoner i stammen refererer til fjerning/innsetting av basepar. For å lage slike mutasjoner brukes oligonukleotid-syntese til å bygge en lang ssDNA, samt en kort ssDNA som baseparer til 3'-enden av den lange og fungerer som en primer i dannelse av dsDNA. Nedenfor vises et eksempel på en slik DNA, hvor innsatte baser er markert med fet tekst.
3'-TTCGAAAACTGTGCCCACGGGTAGCCAACGTCCAGACGTGGTTAGCCATGGGCCAAAAACCTAGG-5'
De manglende nukleotidene fylles inn med DNA polymerase I (Klenow) slik at det dannes en komplett dsDNA. Nedenfor vises DNA etter fyll-inn, hvor UGA-kodonet (TGA i DNA) er understreket og selenocysteine insertion sequence (SECIS) er markert med fet tekst.
3'-TTCGAAAACTGTGCCCACGGGTAGCCAACGTCCAGACGTGGTTAGCCATGGGCCAAAAACCTAGG-5'
(Hvorfor løkke-mutantene og stamme-mutantene produseres med forskjellige metoder, hvor løkke-mutantene bygges fra fire korte sekvenser og stamme-mutantene bygges fra en kort og en lang sekvens, vet jeg ikke.)
I produksjonen av løkke-mutantene ble T4 DNA polymerase brukt for å fylle inn nukleotider og danne komplette dsDNA, mens i produksjonen av stamme-mutantene benyttes DNA polymerase I (Klenow) for å fylle inn nukleotider. Klenow kan ikke brukes til å fylle inn nukleotider i løkke-mutantene fordi Klenow har evne til å skyve bort DNA foran seg (såkalt "strand displacement"), slik at DNAs double-stranded regioner løsner fra hverandre (se figuren nedenfor). T4 mangler evnen til å skyve bort DNA foran seg og brukes derfor til å fylle inn nukleotider i løkke-mutantene.
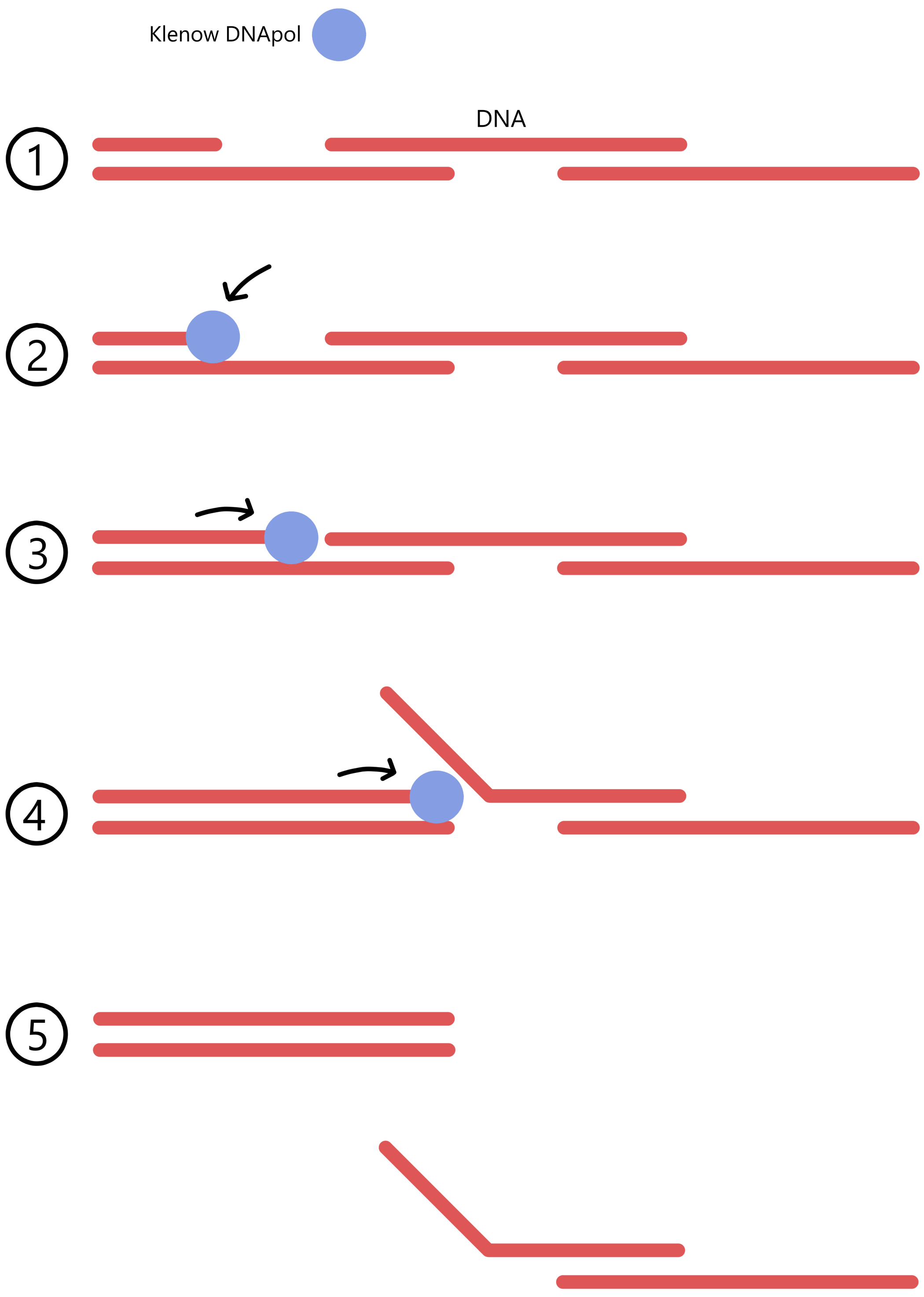
I produksjonen av stamme-mutantene ble DNA polymerase I (Klenow) brukt for å fylle inn nukleotider. (T4 DNA polymerase kunne også ha blitt brukt her. Jeg gjetter at Klenow er billigere enn T4 og at dette er grunnen til at Klenow ble brukt her.)
In both cases the DNAs were cut with HindIII and BamHI at restriction sites provided at their ends and inserted into plasmid pUC9 or pT3T7lac. After screening by sequencing, the wildtype and mutant DNA were inserted into plasmid pSKS106 and rechecked by sequencing.
Alle løkke- og stamme-mutantene ble produsert med en HindIII restriction site 5' for SECIS og en BamHI restriction site 3' for SECIS.
/ \ / \
5'-AAGCTTTTGACACGGCCCATCGGTTGCGGGTCTGCACCAATCGGTCGGTTTTTGGATCC-3'
3'-TTCGAAAACTGTGCCGGGTAGCCAACGCCCAGACGTGGTTAGCCAGCCAAAAACCTAGG-5'
Plasmid pSKS106 inneholder et lacZ-gen hvor en polylinker har blitt satt inn mellom lacZ's femte og sjette kodon (ifølge Heider's referanse: Shapira 1983). SECIS-mutantene settes inn i polylinkerne ved bruk av restriksjonsnukleasene HindIII og BamHI. (Jeg vet ikke hvorfor mutantene settes inn i plasmid pUC9/pT3T7lac før de kuttes ut og settes inn i plasmid pSKS106.)
lacZ med innsatt SECIS-mutant vil transkripteres fra lac-promoteren og oversettes fra lacZ’s start-kodon. 5’-delen av lacZ, som inkluderer SECIS-mutanten, ble sekvensert for å verifisere at riktig DNA-sekvens har blitt satt inn på riktig sted i lacZ.
Syntese av tilfeldige SECIS-mutanter i Klug, Huttenhofer, Kromayer, Famulok (1997): In vitro and in vivo characterization of novel mRNA motifs that bind special elongation factor SelB
(Denne seksjonen krever forståelse av oligonukleotid-syntese.)
Til Klug's forsøk ble det produsert SECIS-RNA med en tilfeldig mutasjonsrate på 30% ("30% degenerate insert", som han selv kaller det). Å lage tilfeldig muterte RNA med oligonukleotid-syntese er enkelt. Først lages tilfeldig muterte DNA, og deretter transkripteres RNA fra disse DNA.
Her er en DNA med en villtype SECIS:
Dersom denne sekvensen skal produseres med oligonukleotid-syntese starter man med å bruke thymin-nukleotider, deretter guanin, så guanin igjen, så cytosin, så thymin, ... slik at en får sekvensen 3'-TGGCT... (Ved oligonukleotid-syntese bygges DNA fra 3'-enden mot 5'-enden.)
Dersom det derimot skal produseres en SECIS med en tilfeldig mutasjonsrate på 30% starter man med å tilsette en blanding av nukleotider hvor det er 70% thymin-nukleotider og 30% andre nukleotider (10% adeniner, 10% cytosiner, 10% guaniner). Deretter tilsetter man en blanding av 70% guanin og 30% andre nukleotider. Så 70% guanin og 30% andre nukleotider, så 70% cytosin og 30% andre nukleotider, så 70% thymin og 30% andre nukleotider, ...
Det lages da et stort antall ulike DNA-sekvenser, hvor hver base i en DNA har 70% sannsynlighet for å matche SECIS-villtypen og 30% sannsynlighet for å være mutert. Som nevnt i seksjonen om Klug 1997 så betyr dette at den gjennomsnittlige DNA vil ha 12 muterte baser i SECIS (39 SECIS-baser × 0.3 mutasjonsrate ≈ 12 muterte baser), men siden prosessen er tilfeldig vil mange SECIS ha flere eller færre enn 12 mutasjoner.
De DNA som produseres av Klug inneholder ikke kun SECIS, de inneholder blant annet også en T7-promoter, som sammen med T7 RNA polymerase brukes for å transkriptere DNA til RNA. De transkripterte SECIS-RNA blandes med SelB-proteiner for å undersøke hvilke SECIS-mutanter som binder SelB.
Kilder
Kromayer, Wilting, Tormay, Bock
(1996): Domain Structure of the Prokaryotic Selenocysteine-specific Elongation Factor SelB. Betalt artikkel
Shapira, Chou, Richaud, Casadaban
(1983): New versatile plasmid vectors for expression of hybrid proteins coded by a cloned gene fused to lacZ gene sequences encoding an enzymatically active carboxy-terminal portion of beta-galactosidase. Betalt artikkel
Smeal, Schmitt, Pereira, Prasad, Fisk
(2016): Simulation of the M13 life cycle I: Assembly of a genetically-structured deterministic chemical kinetic simulation. Fri artikkel